2 Department of Biochemistry and School of Biological Sciences University of Utah, Salt Lake City, UT, USA
Transmission electron microscopy (TEM) is used extensively to study the nematode C. elegans. A typical specimen preparation for TEM involves chemical fixing followed by consecutive changes of reagents and embedding in plastic before thin sectioning. To handle this extremely small animal (~ 1 mm in length and 50 μm in diameter for adults), the most popular method is to immerse chemically fixed worms in warm agarose (Hall et al., 2012). Upon cooling, the solidified block is then trimmed to a maneuverable size. The goal and merit of this method is to avoid losing precious specimens during solution change and specimen transfer, as well as to facilitate desired body orientation prior to sectioning. Here we report the use of hollow fibers in lieu of agarose to achieve the same goal.
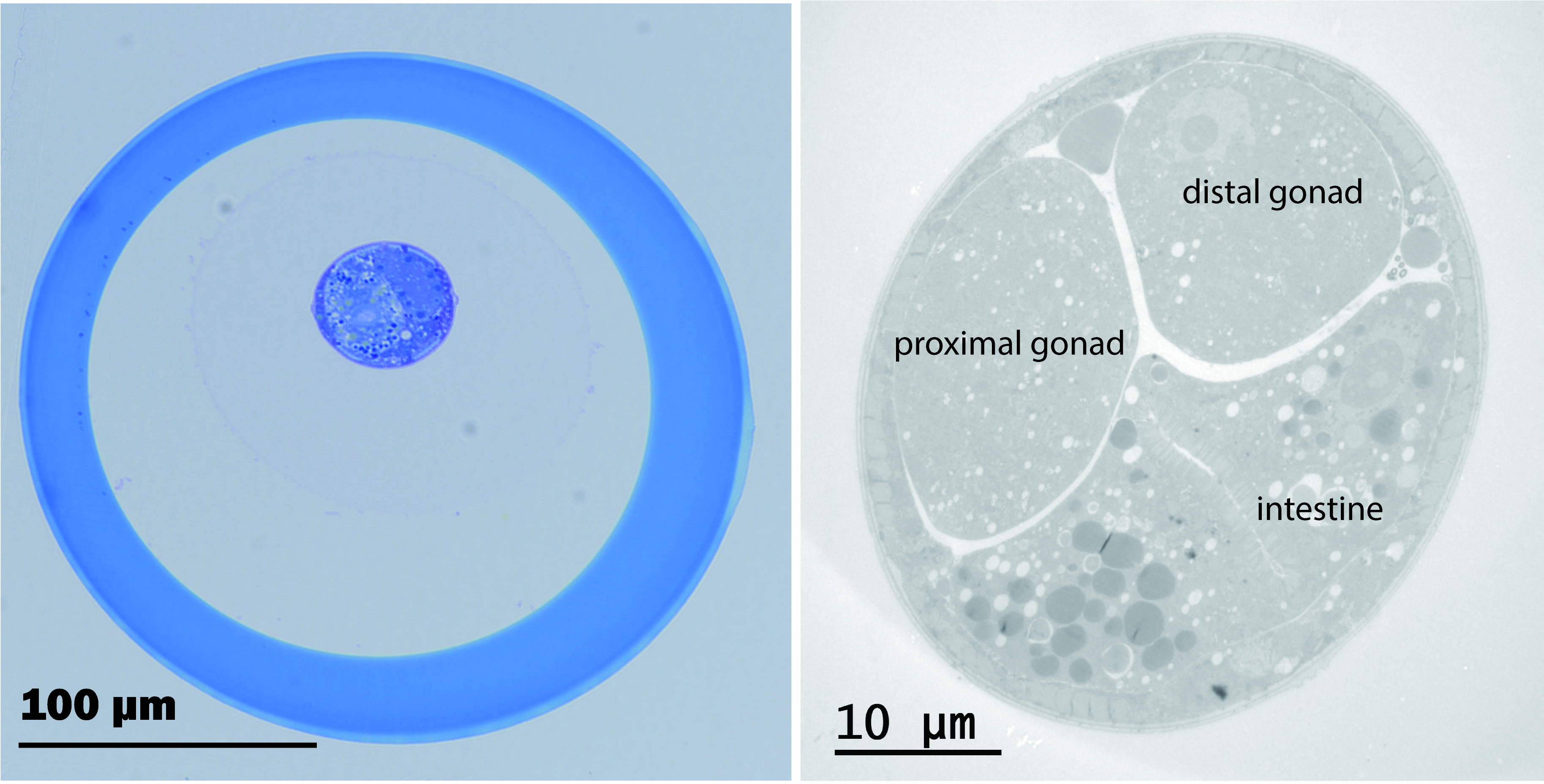
We used transparent hollow fibers from a hemodialyzer (Figure 1A) to encapsulate the worm. Each fiber has an inner diameter of 175 μm and wall thickness of 25 μm. For easy manipulation of fibers, an insect pin1 of 100 μm in diameter is inserted into the lumen of the fiber (Figure 1B, Video 1). To encapsulate, a piece of appropriately fixed worm specimen is transferred to a ~ 3μl drop of buffer rinse and then one end of the hollow fiber is submerged into the drop. With capillary action, the specimen is drawn up into the fiber (Video 1). This loaded fiber is handled with tweezers and processed normally as for tissue samples2. At the end of resin infiltration, excess length of the fiber is trimmed off and the worm can be oriented as desired in a horizontal mold (Figure 1C). During curing, the specimen will sink into the bottom of the mold due to gravity. The fiber gives sufficient leeway (at least 25 μm) at the bottom of the block for trimming (Figure 1D). Therefore, pre-filling the mold or re-embedding the specimen is not necessary (Mulcahy et al., 2018, Muller-Reichert et al., 2003). This greatly expedites the thin-sectioning process. Figure 2A shows that the hollow fiber, made of ethylene vinyl alcohol copolymer, remains intact after exposure to osmium, uranyl acetate, alcohol, acetone and resin Embed 812. Fine TEM images of the worm (Figure 2B) indicate that the hollow fiber, with its molecular weight cutoff at ~30 kDa, allows for penetration of these chemicals. We have also successfully encapsulated the worm specimens by simple capillary action while they were soaked in 1:1 mixture of resin and acetone. Therefore, encapsulation is applicable to samples that are prepared by other means, for example by high pressure freezing and freeze-substitution with organic solvents.
1 A Minutiens insect pin is used (size 0.10, Austerilitz Insect Pins®). Individual pins are about 12 mm in length, 0.1 mm in diameter at the shaft, and 0.0125 mm in diameter at the tip (Figure 1). For easy manipulation of the pin, one end is heat annealed to a p10 pipette tip (Video 1).
2 We use ~2.5 cm length of the hollow fiber for encapsulation and place the loaded fiber into 2 ml microfuge tube for solution changes. If the worm is situated in the middle of the fiber, there is no worry of losing the specimen during processing. However, if the hollow fiber were to be trimmed shorter, both ends need to be sealed either by squeezing with forceps (Video 1) or by heating with hot platinum wire to prevent escape of the specimen.

References
Hall DH, Hartwieg E, and Nguyen KC (2012). Modern electron microscopy methods for C. elegans. Methods Cell Biol. 107, 93-149.

Mulcahy B, Witvliet D,Holmyard D,Mitchell J,Chisholm AD,Meirovitch Y, Samuel ADT, and Zhen M (2018). A pipeline for volume electron microscopy of the Caenorhabditis elegans nervous system.
Müller-Reichert T, Hohenberg H, O'Toole E T and McDonald K. (2003). Cryoimmobilization and three-dimensional visualization of C. elegans ultrastructure. J. Microsc. 212(Pt 1), 71-80.
Single worm transfer with a pin hook
Breeding worms in the laboratory typically involves transferring individual animals from one plate to another. This is usually done with a platinum wire, or sometimes a hair (e.g. eyelash or eyebrow). The wire and hair are both tiny rod-like segments and are generally mounted to a pipette or a stick for easy handling. Hairs are useful only for small larvae transfer. With larger animals, the hair is either too weak to lift them up, or too stiff that the creatures suddenly straighten out and dart away upon touching. A tip-flattened platinum wire a.k.a. “the worm pick” can hold heavier animals. However, they often linger around the pick and refuse to land on the plate. Here we introduce an angle bent pointed hook made of insect pin for single worm transfer that is applicable to all developmental stages of worm from L1 to adult gravid.
The smallest-sized insect pin1 is used. These pins are sold in a package of 500 and are more affordable than platinum wires. Individual pins are about 12 mm in length, 0.1 mm in diameter at the shaft, and 0.0125 mm in diameter at the tip. Although small, its pointed end is not sharp as to poke holes in the worms and kill them. The pin, being made of stainless steel, can be bent to a fixed angle. To make a hook, we clamp the pin at about 0.3 mm from the pointed end with tweezers, and then pull the shaft to 90° angle. This angle-bent pointed hook, like platinum wire and hair, is then annealed to a pipette tip. An easy way is to pass a p10 polypropylene pipette tip over an alcohol flame and quickly insert the blunt end of the insect pin into it. When in use, the pick is sterilized by dipping in 70% alcohol and let air-dry. Coating the pick with sticky bacteria is not necessary. Because its tip is L-shaped, the pick can be lowered vertically onto plate without poking a hole in agar. The tapered end, now runs parallel to the agar surface, is glided under the worm to shovel it up2. A sudden prodding will elicit an abrupt response; however, the worm remains on the plate locally. For younger animals a couple swipes may be needed to pick them up. For large animals, if the maneuver is slow enough, the worm will tangle with the pick (as if mistaken it for another worm) and be elevated. Once lifted, the worm dangles precariously from the hook. The worm will crawl away immediately when the new plate surface is reached.
1 Minutiens, Austerilitz Insect Pins®
2 Orient the pointed tip at 12 o’clock direction so that the stroke, from lower left to upper right (for right-handed person), is an upward moment. Doing so would avoid poking holes in agar.
Figures


“N2 male” is a long-lived fln-2 mutant
To distinguish wild-type and mutant genes, one needs to define a wild-type strain. To this end, we worm breeders use the convention that N2 is the wild type. Now, when people order N2 from the Caenorhabditis Genetics Center, they usually request a hermaphrodite stock, but there is also a male stock available (strain: “N2 male”); for convenience, we will refer to these two lines as N2H and N2M. A problem here is that these lines are not genetically identical: N2M hermaphrodites are longer lived (+11% median lifespan) (Gems and Riddle, 2000). So which is wild type, N2H or N2M? This is a problem, and we have been looking into it.
Under standard culture conditions N2 hermaphrodites exhibit two forms of death, with either a swollen or an atrophied pharynx (P and p death, respectively). P death occurs earlier and results from bacterial infection, facilitated by mechanical senescence of the pharynx due to the high, wild-type pumping rate (Zhao et al., 2017). We have found that the greater lifespan of N2M hermaphrodites is due to lower P death frequency (and therefore reduced early mortality).
This turns out to be due to a single recessive mutation in the X-linked gene fln-2 (filamin) in N2M, as revealed initially by Variant Discovery Mapping. fln-2(ot611) is a nonsense allele resulting from a C to A transversion, creating a stop codon at Y800 in the FLN-2A isoform. Thus, N2H is wild type and N2M is mutant. This conclusion is confirmed by examination of the fln-2 sequence in multiple C. elegans wild isolates (Cook et al., 2017). It would therefore be advisable to discontinue the use of N2M (“N2 male”).
Although people mainly use N2H as their wild type, N2M is sometimes used for strain construction and backcrossing. To get an idea of the prevalence of fln-2(ot611) we checked a sample of strains in our collection and found fln-2(ot611) in 23/50, particularly in strains generated by the C. elegans Gene Knockout Project and the C. elegans Expression Project.
Variation at the fln-2 locus is a potential confounding variable, especially in studies of lifespan genetics. We have so far found it to confound the effects on lifespan of alteration of eat-2, sir-2.1 and daf-12. For this reason we advise the use of routine checks of fln-2 genotype in studies of the genetics of lifespan. Here is information about how to do this.
The relevant fln-2 sequence information is:
Wild-type fln-2: GGCGCTGGTCAATA[C]AAAATCCACGTTCTT
fln-2(ot611): GGCGCTGGTCAATA[A]AAAATCCACGTTCTT with a C to A change in the bracket.
To genotype fln-2, we used the following primers to PCR amplify the region containing the mutation: forward 5’-GGTGTTCGATTCTGGTCTGG; reverse 5’-ACATCGACGAGAAGACAACAC. The PCR product can be sequenced using the primer 5’-TGTACCCAGAAATTGACAAGATAC.
Allele-specific PCR can also be used to discriminate between the alleles, using the following primers: wild-type-specific forward 5′-taccattccgagcttattgattgttacctGGACGGCGCTGGTCCATAC-3′; ot611-specific forward 5′-GGACGGCGCTGGTCTATAA-3′; reverse 5′-ATCGCATGAACCATAAATGATG-3′. Each forward primer contains an additional mismatch to make it different from both the template and the other forward primer to ensure allele specificity (Neagu and Maier, 2011). The wild-type PCR product is 30bp longer than the mutant product, and can be readily distinguished on a 2% agarose gel.
References
Cook D, Zdraljevic S, Roberts J, and Andersen E. (2017). CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Research. 45, D650-D657.
Gems D and Riddle DL. (2000). Defining wild-type life span in Caenorhabditis elegans. J. Gerontol. A Biol. Sci. Med. Sci. 55, B215-B219.
Neagu A and Maier W. (2011). snPCR for reliable one-step genotyping of single nucleotide differences. The Worm Breeder’s Gazette 19, 1.
Zhao Y, Gilliat AF, Ziehm M, Turmaine M, Wang H, Ezcurra M, Yang C, Phillips G, McBay D, Zhang WB, Partridge L, Pincus Z, and Gems D. (2017). Two forms of death in ageing Caenorhabditis elegans. Nature Comm. 8, 15458.
Review of 3rd Parasitic Nematodes: Bridging the Divide workshop
2 Department of Molecular, Cell, and Developmental Biology, University of California, Santa Cruz, USA
Soil-dwelling helminths such as hookworms, whipworms, and Ascaris infect hundreds of millions of people globally, and also infect livestock and crops. Dick Davis (University of Colorado School of Medicine) gave an overview of Ascaris, highlighting resources, tools, and key questions in this system. Two fascinating features of the system he discussed were somatic DNA elimination, in which 15% of the genome is removed in somatic cells during embryogenesis, and early zygotic genome transcription commencing remarkably early (immediately following fertilization). Elissa Hallem (UCLA) discussed parasitic nematode sensory behaviors with respect to host seeking. Using both Strongyloides nematodes and C. elegans, her group is exploring the neurobiology of how parasites find their hosts. Tiffany Baiocchi, a PhD student with Adler Dillman (University of California, Riverside) presented her work on novel odorants produced from nematode-parasitized insect cadavers and the behavioral responses this odorant triggers in both free-living and parasitic nematodes. Jonathan Ewbank (Centre d’Immunologie de Marseille- Luminy) gave a lightning talk on assembling the Nippostrongyloides brasieliensis genome and dealing with complex repeats; this nematode is a parasite of rodents and a useful model for human hookworm infection.
Two experts discussed filarial parasites, a group of nematodes that cause lymphatic filariasis and onchocerciasis. Several of these species carry obligate endosymbiotic bacteria (Wolbachia), which is an attractive therapeutic target. Malina Bakowski (California Institute for Biomedical Research) discussed her high-throughput screens to identify potent anti-Wolbachia molecules. Sara Lustigman (New York Blood Center) presented on the role of “omics” and molecular tools in Brugia species efforts to eliminate nematodes that cause the human disease filariasis. Conor Caffrey (University of California, San Diego) also presented a high-content screening system for flatworms which cause schistosomiasis.
Adrian Wolstenholme (University of Georgia) discussed the problem of resistance to anthelminthics and the mechanisms of resistance. His group uses a combination of heterologous studies in C. elegans and work in parasites such as Brugia malayi and Haemonchus contortus to explore potential mechanisms of resistance to drugs such as ivermectin and diethylcarbamizine. Nidhi Sharma, a Ph.D student with John Gilleard (University of Calgary) presented a lightning talk on the role of UDP-glucuronosyltransferase (UGT) enzymes in benzimidazole drug biotransformation by nematodes. Keeping with the theme of glycosylation, Patricia Berninsone (University of Nevada, Reno) gave a lightning talk on her identification of nematode phosphorylcholine-modified N-glycoproteins using C. elegans.
This was the third time that the workshop was held and it brought together 77 participants to explore a range of approaches and systems to explore parasitic nematode biology and to define areas where C. elegans researchers can make significant impact. Going forward, we aim to integrate the workshop into regular program of the worm meeting as having it beforehand limits the ability of some researchers attend. Look for us at the 2019 Worm Meeting!
Troubleshooting the positive butanone associative memory assay
2 LSI Genomics & Dept. of Molecular Biology, Princeton University, Princeton NJ, USA
Learning assays for C. elegans are known to be challenging due to the fragility of the phenotype, hence, the ease by which it can be disturbed. For several months, our teams at KU Leuven struggled to implement the Murphy lab’s short-term associative memory assay, failing to obtain proper control data despite having implemented other learning assays without noteworthy incidents. We are aware that others in the field have struggled with this assay as well.
To address this issue, the Murphy lab supported us in troubleshooting their protocol both in Belgium as well as during a stay at their lab in the USA. We discovered several small and seemingly trivial differences which showed to be of major importance for the success of this assay. These observations allowed us to successfully perform the assay in Belgium, where it had not worked previously.
In order to enable others to smoothly implement this assay in their labs, the updated protocol is described on the Murphy and Schoofs labs’ webpages.
Weblink Murphy lab: http://www.molbio1.princeton.edu/labs/murphy/protocols.html
Weblink Schoofs lab: https://bio.kuleuven.be/df/ls/research/positive-butanone-associative-memory-assay