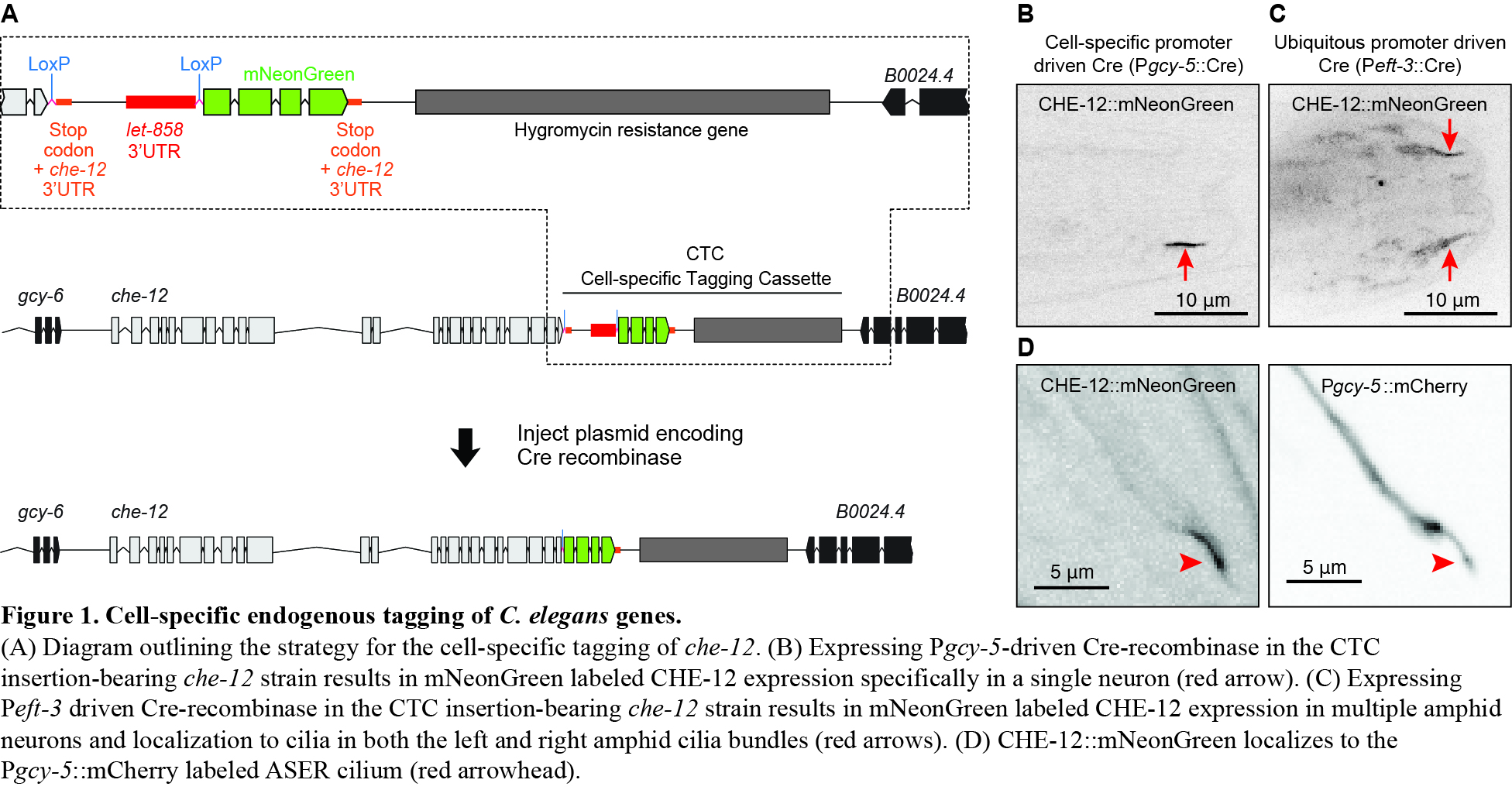
If possible, we use the native CRISPR/Cas9 system, which relies on Cas9 protein with both a universal RNA (tracrRNA) and a specific guide RNA (crRNA). These RNAs are bought from Dharmacon, and the same tracrRNA can be used for every injection. Although commercial Cas9 is also available, it is expensive. Thus, Paix and Seydoux developed a protocol for Cas9 production in E. coli [4]. We modified their procedure to simplify the Cas9 purification. Briefly, the plasmid NM2973 was transformed into NEB Nico-21 competent cells (#C2529H), and Cas9 expression was induced by adding IPTG to the culture media. We purified Cas9 under native conditions, using a Qiagen Fast-Start Kit (#30600). Following instructions, the bacterial pellet was resuspended in 10 ml of lysis buffer, and cooled on ice for 30 minutes. The cell lysate supernatant flowed through the Fast-Start Ni-NTA column by gravity, and 4 ml of washing buffer was used to wash the column twice. Next, Cas9 was eluted with 2 ml of elution buffer. Finally, the Cas9 eluate was centrifuged with a centricon filter (#UFC905024) to concentrate the protein. We obtained 2.5 mg of Cas9 from 1L of culture, and made 2 µl aliquots at a concentration of 10 µg/µl for storage at -80°C.
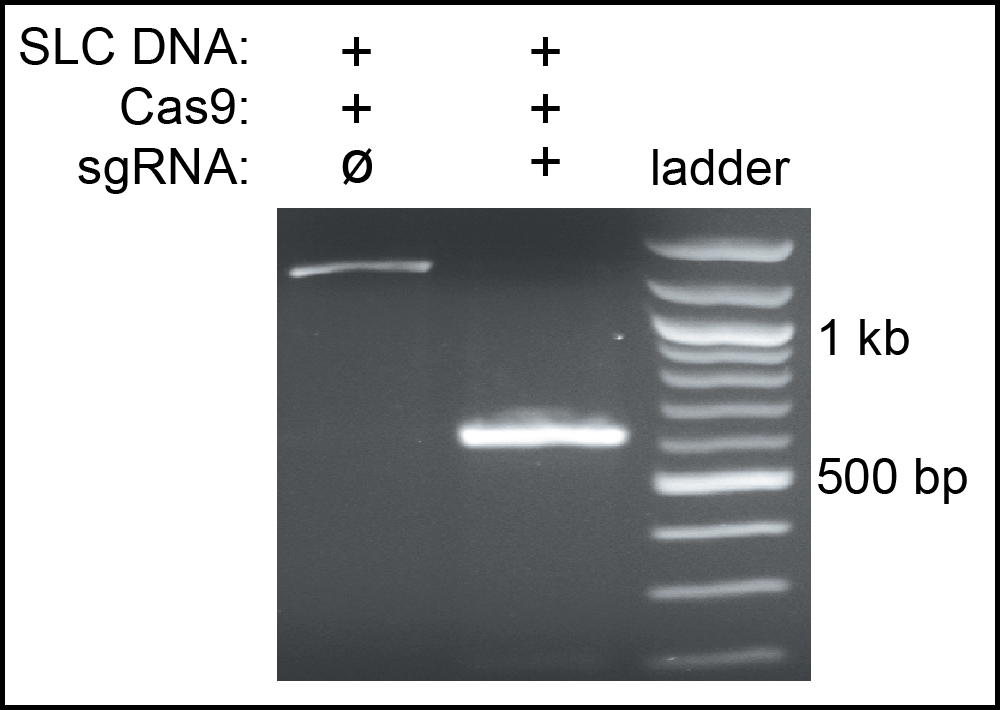
Home-made Cas9 was tested to measure it efficiency. First, we performed a cleavage assay to confirm that it had endonuclease activity. A mixture of Cas9, mouse SLC guide RNA and a PCR fragment from the SLC gene were digested at 37°C for 1 hour. The result showed that our Cas9 completely cleaved the SLC fragment as shown in Figure 1. Second, we used the Cas9 to knockout the cbr-dpy-5 gene in vivo. The synthetic cbr-dpy-5 crRNA and tracrRNA were incubated with Cas9 at 37°C for 15 minutes, and microinjected into the gonads of C. briggase hermaphrodites. Among all the F1 progeny, 25% produced homozygous Dpy children. A similar injection using commercial Cas9 (PNA Bio.) gave 18% affected F1s. Sequencing confirmed that the cbr-dpy-5 mutation was a 51 bp deletion near the PAM site of its target. Thus, home-made Cas9 has high efficiency and allows rapid and inexpensive gene editing.
If the desired target lacks a nearby 3’-GGnGG, TALENs can be used for precise gene editing. Since the only constraint for TALEN binding is that the first nucleotide in each target sequence is a T, sites exist almost everywhere in the genome. The plasmids to synthesize a pair of TALEN mRNAs can easily be built in a week, using a procedure developed by Cermak et al [1], with the destination vector pRE189 for expressing nematode mRNAs [5]. A Golden Gate TALEN kit is available from Addgene (#1000000024) and mRNA kits from Ambion (#AM1344, #AM1908).
Acknowledgments: We thank Dr. Geraldine Seydoux for the NM2973 plasmid. This work was supported by NIH grant to Dr. Ronald E Ellis.
Figures
References
[1] Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, and Voytas DF. (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39(12):1-11 
[2] Dickinson DJ, And Goldstein B. (2016) CRISPR-based methods for Caenorhabditis elegans genome engineering. Genetics. 202(3):885-901
[3] Farbound B, and MeyerBJ. (2015) Dramatic enhancement of genome editing by CRISPR-Cas9 through improved guide RNA design. Genetics. 199(4):959-971
[4] Paix A, Kolkmann A, Rasoloson D, and Seydoux G. (2015) High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics. 201(1):47-54
[5] Wei Q, Shen Y, Chen X. Shifman Y. and Ellis RE. (2014) Rapid creation of forward genetics tools for C. briggase using TALENs: lessons from nonmodel organisms. Mol. Biol. Evol. 31(2):468-473