Correspondence to: Greg Nelson (gnelson@dominion.llumc.edu)

View the full size version.
View the full size version.
Beginning with this issue, we are introducing three new types of content that we hope will make The Worm Breeder’s Gazette more useful to you, the community of nematode researchers. The Gazette has traditionally been a forum for the dissemination of unpublished data and methods, and we plan to continue presenting this type of content. However, the Gazette is also a community newsletter, and we would like to enhance this function.
1). In this and subsequent issues, we will be including author-written summaries of new or improved methods from published papers. These articles, tagged ‘Highlighted publication’, are intended to enhance the visibility of papers published as methods as well as technological advances contained in research papers.
2). Several members of the Editorial Board of WormBook have contributed brief lists of notable recent papers in their areas of expertise.
3). You will also find announcements describing newly established worm labs and their research interests. Subsequent issues may also include announcements of relocating labs, upcoming meetings and promotions and awards given to worm researchers.
As always, we welcome your comments and suggestions concerning The Worm Breeder’s Gazette.
We have created a library of 2,000 mutagenized C. elegans strains, each sequenced to an average depth of 15X to reveal most mutations. The library contains over 700,000 single nucleotide variants (SNVs) with, on average, 8 non-synonymous changes per gene. We generated the library using the mutagens EMS, ENU or a cocktail of EMS plus ENU. F1 populations were screened in nicotine for animals heterozygous for unc-22 mutations to ensure effectiveness of the mutagen. F2 populations were screened again to select non-unc-22 animals, and the resulting lines were selfed for a further eight generations to drive all genomic regions toward homozygosity. Whole-genome sequencing was done with paired-end reads on Illumina GAII or Hi-Seq machines using size-selected and molecularly bar-coded DNAs. Reads were aligned using Phaster (P. Green, unpublished) and SNVs were called using SamTools and custom filters. Indels and rearrangements were identified with custom tools.
Analysis of the data from the first 1,794 strains has yielded 705,748 SNPs in 20,066 genes (averaging 390 per strain). These include 159,338 non-synonymous changes in 19,449 genes (eight new alleles per gene). Of these mutations, 9,829 are knockouts (nonsense or spicing defects) in 6,774 genes, for an average of more than four per strain. Based on read numbers, the rDNA repeat copy number is surprisingly variable, with some strains having fewer than 60 copies and a few having more than 150. We have supplemented these mutagenized strains with 40 natural isolates to recover an additional 500,000 mutations. The mutation data for the first 600 mutated strains have been deposited in WormBase, with the rest of the data in process. A separate website allows direct queries of the data (http://genome.sfu.ca/mmp/). Nearly all of the 2,000 individual strains are available from the Caenorhabditis Genetics Center. We are currently building frozen kits containing all the strains in 96-well arrays, allowing parallel experimentation on a wide spectrum of mutant genes. The resource should provide rapid access to multiple mutations in any gene of interest as well as allow investigation of gene-gene interactions.
WormWiring is intended to supplant the now dated The Male Wiring Project. In addition to asthetic and navigational updates we have added several new features. Compared to the previous static neuron map images, our new neuron maps are loaded on the fly with the ability for the user to select the view, types of synapse displayed, synapse labels, and synaptic weight for their neuron of interest (Fig. 1). We have developed a legible and informative new visual representation of neuronal synaptic partners as well as the identity of partners involved at individual synapses (Fig. 2). For those interested in neural network structure we have supplied an adjacency matrix browser sortable by neuron name, in degree, out degree, in strength, out strength, page rank, and neuron class (Fig. 3).
Currently WormWiring contains the connectivity data seen at The Male Wiring Project, which constitutes the reconstruction of the posterior nervous system of an adult male C. elegans (Jarrell et al., 2012). We are currently reconstructing the anterior nervous system of an adult male and will make these connectivity data available on WormWiring within several months.
In addition to male C. elegans animals, we have revisited the historical EM data used for The Mind of a Worm (White et al., 1986). Our reconstruction encompasses the anterior and posterior nervous systems and includes description of over 6000 synapses. Our new data will include X,Y,Z coordinates of synapses and neuronal processes, improved characterization of synaptic weight, as well as clarification of neuromuscular junction synaptic partners. Again, these data will be made available on WormWiring soon. Connectomics projects that are planned or ongoing include: L1, L2, L4, and dauer animals. We look forward to hosting additional connectivity data from other laboratories.
We encourage other labs to undertake their own reconstructions with our help using our image annotation software, Elegance. There is likely to be interest in reconstructing legacy electron micrographs found by browsing WormImage for developmental stage or genotype of interest. To date there are 147 and counting different image series hosted, including 70 mutant and 77 wild-type animals of all developmental stages including dauer all waiting to be further analyzed.
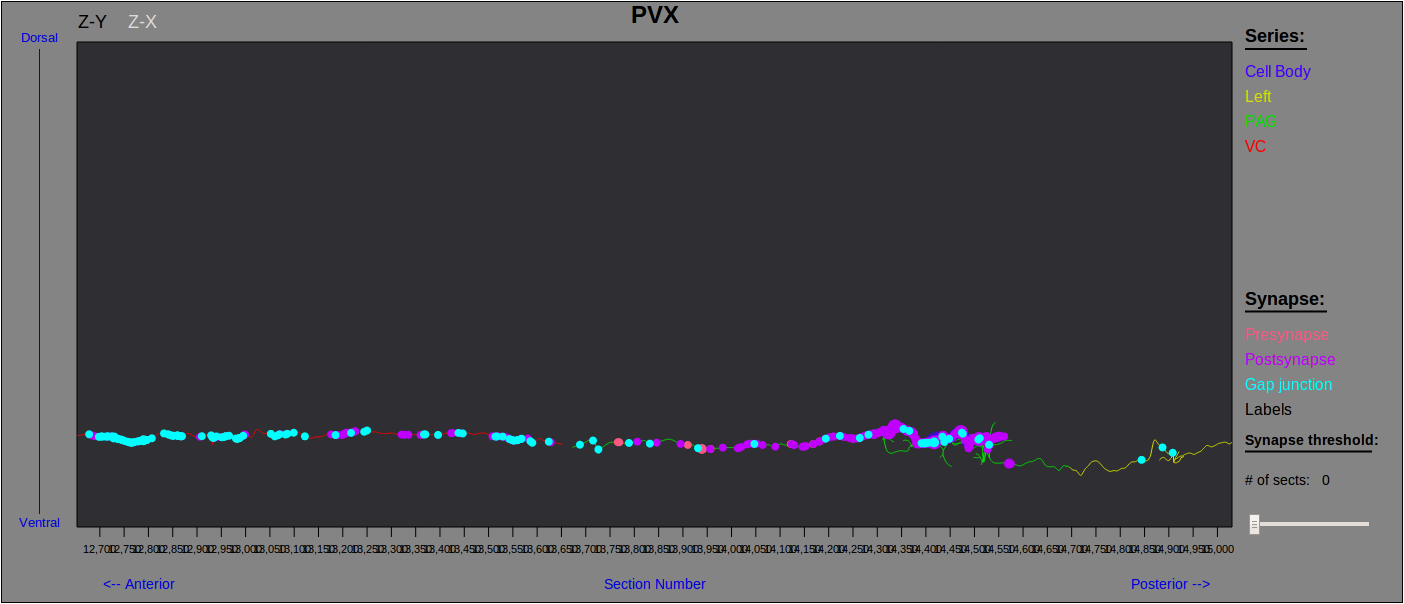
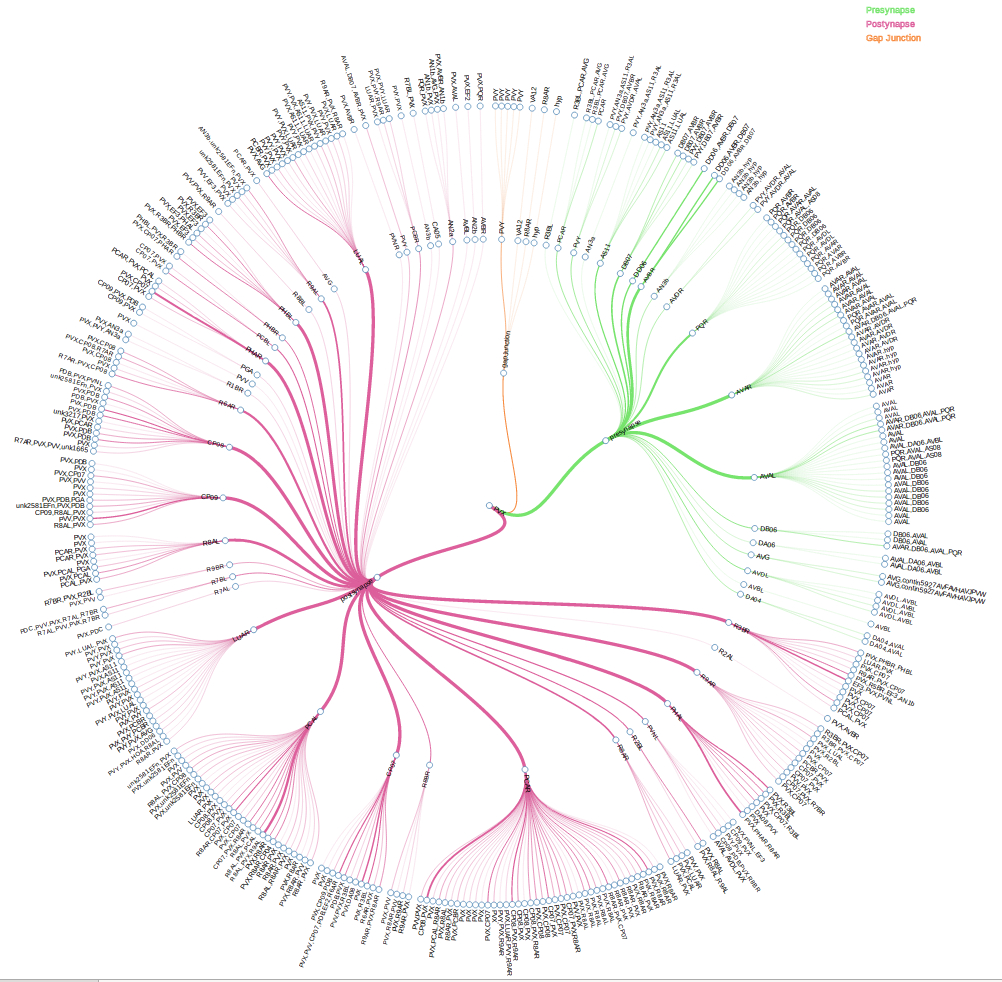

Jarrell et al., 2012. Science. In press.
Male Wiring Project, Albert Einstein College of Medicine, website:
http://worms.aecom.yu.edu/PHP/male_wiring_project.php
The WormWiring Project, Albert Einstein College of Medicine, website:
http://www.wormwiring.org
White JG, Southgate E, Thomson JN, and Brenner S. (1986). The structure of the nervous system of Caenorhabditis elegans. Philosophical Transactions of the Royal Society, Series B Biological Sciences 314, 1-340. 
Oliver Hobert and his colleagues have pioneered the use of whole-genome sequencing (WGS) to identify lesions in C. elegans mutants, and they have produced the MAQGene software pipeline to analyze this data (Sarin et al., 2008) (http://maqweb.sourceforge.net). While MAQGene is excellent, it runs on Linux operating systems and requires a MySQL server, and these requirements are currently beyond our (and perhaps other C. elegans researchers) computer capabilities.
We are using WGS to identify a mutation cu13 that enhances the lethal phenotype of a hypomorphic tbx-2 mutant. Illumina sequencing was used to sequence genomic DNA of several strains that have the cu13 or wild-type alleles. Freely available software packages were used to align sequence reads with the reference C. elegans genome, identify variants, and annotate these variants with predicted effect on gene function. These analyses identified thousands of variants in each sequenced genome, and Microsoft Access was used to sort and compare variants in each genome. A small number of candidate lesions for cu13 were identified, and we are currently determining which of these causes the mutant phenotype. This approach is feasible for novices like us using a desktop computer and fairly rudimentary skills with the command line interface, and we thought others in the C. elegans community might be interested in trying this for themselves. The software packages generally have manuals and tutorials available, and we relied on these heavily.
Sequence alignment: Bowtie 2 was used to index the C. elegans reference genome and to align our fastq sequencing reads to this reference (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). Bowtie 2 is an ultrafast aligner that outputs a SAM (Sequence Alignment/Map) used in subsequent analyses (Langmead and Salzberg, 2012 PMID 22388286), although Bowtie 2 may be less sensitive than the MAQ aligner used in MAQGene (Nielsen et al., 2011).
Variant identification: The SAMtools software package was used to identify variants and call genotypes based on SAM alignment files (http://samtools.sourceforge.net/) (Li et al., 2009). SAM files were initially converted to their binary equivalent BAM format and sorted using ‘samtools view’ and ‘samtools sort’ commands. Information regarding sequence quality and possible genotype was calculated using the ‘samtools mpileup’ command and stored in the BCF file format. Variants were called and written to a VCF (Variant Call Format) file using the ‘bcftools view’ and ‘vcfutils.pl’ commands. VCF is a widely used text file format storing information regarding variant position and sequence, sequence quality, and predicted genotype.
Variant annotation: C. elegans genome annotations were retrieved from the UCSC Genome Browser Annotation Database using the Perl-based software package ANNOVAR (http://www.openbioinformatics.org/annovar/) (Wang et al., 2010). ANNOVAR was used to convert our VCF files to ANNOVAR input files and annotate variants using the ‘perl convert2annovar.pl’ and ‘perl annotate_variation.pl’ commands. ANNOVAR outputs one file annotating all variants indicating the genomic features they hit, and a second file indicating the amino acid changes for exonic variants. For convenience, these files were combined into a single table using Microsoft Access.
Variant and sequence visualization: The Integrative Genomics Viewer (IGV) (http://www.broadinstitute.org/igv/home) was used to visualize variants and the underlying sequence reads (Thorvaldsdottir et al., 2012). Variants called in VCF files and sequence alignments in BAM files can loaded into tracks in the IGV browser and can be rapidly viewed at a wide range of genomic scales.
There are a variety of software options available for each of the steps (Nielsen et al., 2011), and while the software described here works for us, we are evaluating other approaches for each of these steps.
Langmead B, and Salzberg SL. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078-2079.
Nielsen R, Paul JS, Albrechtsen A, and Song YS. (2011). Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443-451.
Sarin S, Prabhu S, O’Meara MM, Pe’er I, and Hobert O. (2008). Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods 5, 865-867.
Thorvaldsdottir H, Robinson JT, and Mesirov JP. (2012). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. April 19 (Epub ahead of print).
{kind=link}