We prepared F1 eggs by basic hypochlorite treatment of EMS-mutagenized eat‑5 parents, grew them to adulthood on HB101, then plated their F2 eggs on DA837, about 100/plate. Screen statistics were as follows:
|
recessive |
dominant |
|||||||||
|
Screen |
F1s |
F2s |
μ1 |
P2 |
N3 |
n4 |
μ1 |
P2 |
N3 |
n4 |
|
1 |
18,000 |
7,840 |
0.11 |
0.10 |
3,700 |
1.06 |
0.33 |
0.28 |
10,000 |
1.17 |
|
2 |
11,500 |
39,000 |
0.85 |
0.57 |
13,000 |
1.48 |
2.5 |
0.92 |
21,000 |
2.8 |
1 Mean number of phenotypically mutant F2s per mutation in the F1,  (recessive) or
(recessive) or  (dominant).
(dominant).
2 Probability that a mutation present in the F1 is detected, P=1-e-μ.
3 Effective number of genomes screened, N=2F1P.
4 Mean number of times a mutation is isolated, if it is found at all, 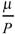 .
.
A mutant line was established from each plate that starved by day 13. In addition, we noticed one plate on which most of the worms were adults rather than L1s and established a line from it (ad1614), even though it didn’t starve until day 16. We thought this mutation might allow escape from the L1 arrest but have other adverse phenotypes that decreased its growth rate.
From these two screens we got a total of 13 sef (suppressor of eat five) mutations. 9 recessive mutations mapped to a single complementation group on LGI, provisionally called sef‑1. sef‑1 mutants had normally synchronized corpus and TB contractions. 3 mutations were dominant, and one of these mapped to LGIII. The remaining mutation, ad1614, was recessive but difficult to map because of the weak phenotype. It looked like something we had seen before. Although corpus and TB were still unsynchronized in eat‑5; ad1614 worms, TB pumping was about twice as fast as in eat‑5. eat‑5; slo‑1 worms have the same phenotype, because M4 synapses onto TB muscle are activated by loss of the K+ channel encoded by slo‑1 (Chiang et al., 2006).
We sequenced the genomes of 9 sef mutant strains: 5 carrying sef‑1 mutations, 3 carrying the dominant sef mutations, and ad1614. Unsurprisingly, the ad1614 mutant had a splice donor mutation in slo‑1. All 5 sef‑1 mutants had mutations in cfi-1 LGI. There were 3 distinct mutations: a splice donor mutation, a splice acceptor mutation, and a nonsense mutation. Based on linked SNPs, these 5 mutations comprise 4 independent events. CFI‑1 is a transcription factor expressed in pharyngeal muscle and other places (Shaham and Bargmann, 2002 ). One of many plausible hypotheses is that in the absence of CFI‑1 an innexin that can replace EAT‑5 is expressed in pharyngeal muscle.
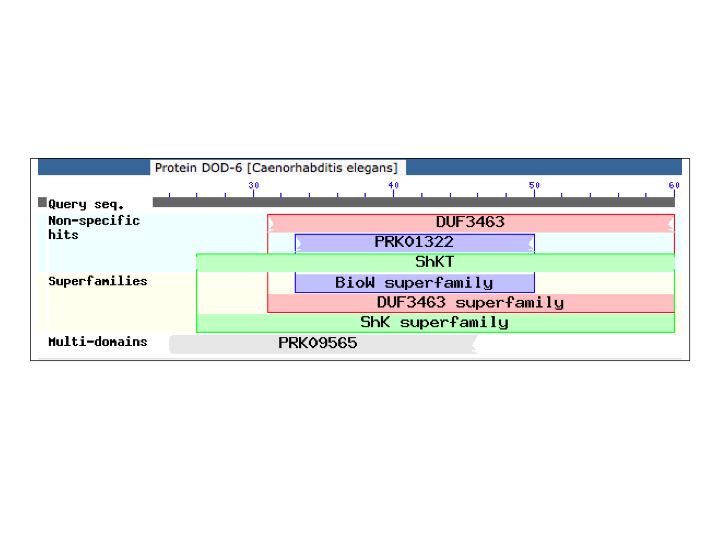
The dominant mutations were most unexpected. All 3 (2 independent) carry the identical A24V mutation in dod‑6 LGIII. Little is known of dod‑6 function. It was identified as a gene downstream of daf‑16, and RNAi slightly decreases lifespan of daf‑2 mutants (Murphy et al., 2003). It encodes a tiny 60 amino acid protein. The N-terminus looks like a signal sequence. The C-terminus is similar to a Metridin-like ShK domain (Figure 1). The eponymous proteins are 33 amino acid sea anemone toxins that block voltage-gated K+ channels. A24 is near the end of the putative signal sequence, 2 amino acids upstream of the toxin domain.
Finding that the same screen yields loss-of-function mutations in a K+ channel and dominant mutations in something that looks like a K+ channel blocker suggests an obvious hypothesis: DOD‑6(A24V) blocks SLO‑1. It is not equivalent to complete loss of SLO‑1, since eat‑5; dod‑6 mutants are healthier and grow faster than eat‑5; slo‑1. The action of DOD‑6(A24V) might be limited to particular times or places, or might be something other than a simple block of all SLO‑1 isoforms, or might have other targets. The hypothesis that DOD‑6(A24V) blocks SLO‑1 makes many experimental predictions, some of which we plan to test.
Figures
References
Chiang JT, Steciuk M, Shtonda B, Avery L. (2006) Evolution of pharyngeal behaviors and neuronal functions in free-living soil nematodes. J Exp Biol 209, 1859-1873. 
Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277-283
Shaham S, Bargmann CI. (2002) Control of neuronal subtype identity by the C. elegans ARID protein CFI-1. Genes Dev 16, 972-983.
Articles submitted to the Worm Breeder's Gazette should not be cited in bibliographies. Material contained here should be treated as personal communication and cited as such only with the consent of the author.
Leave a Reply
You must be logged in to post a comment.