
View the full size version.
View the full size version.
Generating transgenic lines is a challenging task for two reasons: first, there is a bit of a learning curve in mastering the art of microinjection. Second, not every lab has access to the rather expensive paraphernalia required. This is especially true in primarily undergraduate institutions. In fact, the very requirement of having to generate transgenic worms may dissuade such institutes from performing forward genetic screens.
Availability of transgenic lines carrying defined pieces of genomic DNA would circumvent both problems and would be an invaluable tool for the C. elegans community. One can simply cross transgenic lines with the mutant of interest to test for rescue.
Many labs generate several transgenic lines carrying defined piece(s) of genomic DNA for each rescue experiment. Typically, when a researcher X generates several lines, only one rescues the mutant phenotype and rest are either thrown out or kept frozen. Let us assume researcher A finds that the locus of his/her mutant lies within the same region. It would be far more convenient for A to borrow the lines from X than to repeat the same process all over again.
I suggest a system that would facilitate the sharing of transgenic animals carrying defined piece(s) of genomic DNA between labs. First, we should create a website CTGC (Caenorhabditis TransGenics Center). If a ‘donor lab’ has a transgenic line, s/he should visit CTGC website and enter details such as strain name, genotype, description, etc. The submitted information will be stored in a searchable database. The consumer lab can search this database by using key word, such as Cosmid name, which can then be directly requested through CTGC by completing a simple form.
Creating CTGC website is a relatively simple process. However, with the steady demand of transgenic strains from across the globe, many PIs may become reluctant to provide this service, which is quite understandable. Should some transgenic worm become as popular as daf-16 or daf-2 mutant, it is easy to imagine the work-load of the lab of its origin!
So, I suggest a policy in which, by submitting strain information, the donor lab is expected to distribute the strain to only 3 labs. Any subsequent request for the strain will be redirected to the recipient labs. The lab that made the strain won’t be held responsible for distributing the strain indefinitely! It is obligatory that the lab that receives the transgenic strain should ship it forward to at least 3 of the subsequent requests. The strain request will be handled in this way for the rest of the requests.
In order to keep track of the strain distribution, the entire strain request has to be processed through CTGC, which automatically registers the details of all the researchers who received this strain.
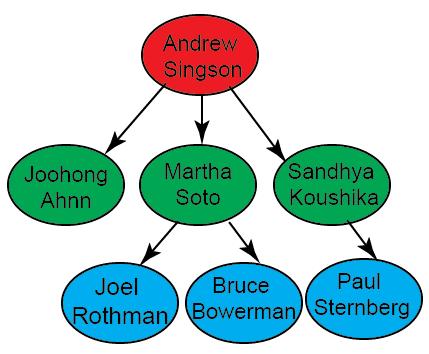
In the hypothetical example (figure 1), the transgenic line is originated from Singson lab. Singson lab has distributed this strain to 3 labs (Ahnn, Soto and Koushika labs). Therefore, the system will no longer show Singson lab as a potential source for this strain. Soto lab and Koushika lab are responsible for distributing this strain to one and two more labs, respectively. Finally, Ahnn, Rothman, Bowerman and Sternberg labs are responsible for distributing this strain to three other labs.
I believe that C. elegans community will be immensely benefited from CTGC and I look forward to your comments and suggestions to improve this concept.
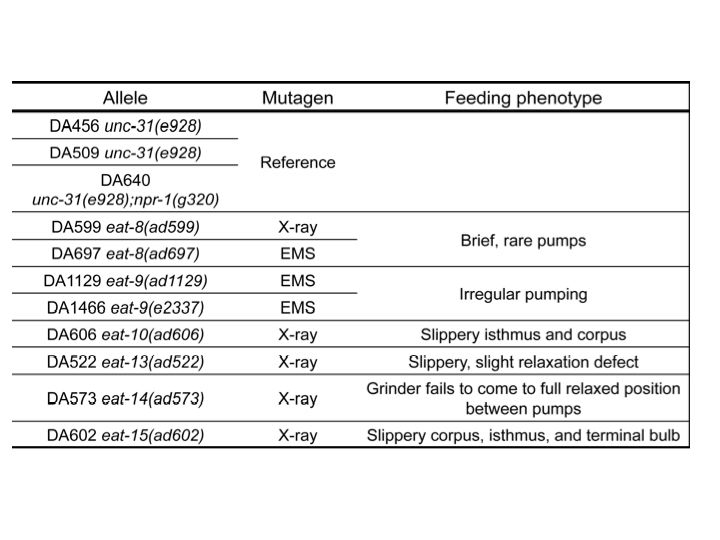
eat-8(ad599) had deletions and eat-8(ad697) had a missense mutation in Y82E9BR.16. This gene encodes a transporter, a member of the solute carrier family 22 superfamily. Mutations affecting similar proteins are associated with increased risk for Crohn’s disease in Homo sapiens (Newman et al., 2005). We are cloning Y82E9BR.16 to verify its identity as eat-8.
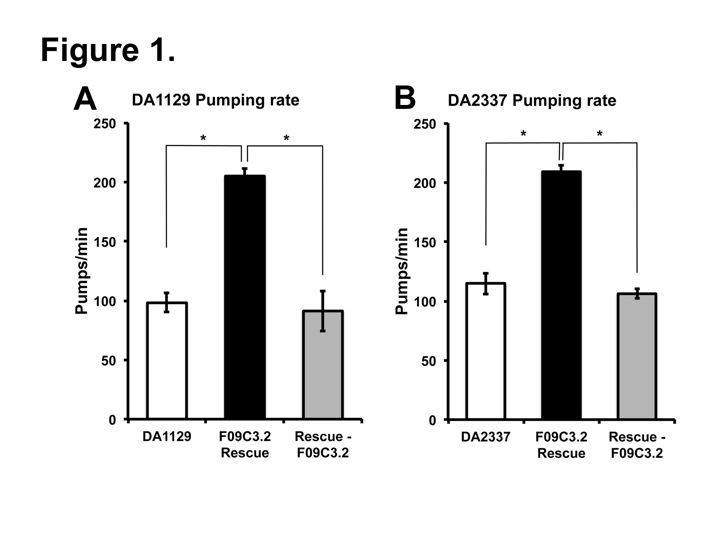
Two independent eat-9 mutants, eat-9(ad1129) and eat-9(e2337), had nonsense mutations at different locations in the coding region of F09C3.2. The eat-9 mutant phenotype was rescued by introducing wild type F09C3.2. When rescued mutants lost wild type F09C3.2, the mutant phenotype was recovered (Fig 1). F09C3.2 encodes a member of the haloacid dehydrogenase (HAD) superfamily (Seifried et al., 2012). We are studying what kind of HAD are related to the irregular pumping phenotype of eat-9.
eat-10 had a frame shift mutation in grld-1, orthologous to human RBM15 and Drosophila NITO, which affects glutamate receptor expression and extends lifespan about 10% by RNAi (Wang et al., 2010; Samuelson et al., 2007). We are comparing the eat-10 mutant phenotype with the known grld-1 mutant phenotypes.
eat-15(ad602) had a deletion in K10D3.4, which encodes a serine protease inhibitor. We cloned and made transgenic worms with this gene and are planning to measure the pumping rate and growth rate and compare the mutant phenotypes.
eat-13(ad522) had a deletion of over 70 kbp, which removed at least 19 genes from R07B1.5 to tsp-21 on chromosome X. We are planning fosmid and cosmid rescue experiments to identify which genes are related to the mutant phenotype. eat-14(ad573) had a 50 kbp deletion which removed at least eight genes from rsd-3 to F54F7.2 on chromosome X. We will also perform fosmid rescue experiments on this mutant. It is of course possible that the mutant phenotypes depend on multiple genes.
In conclusion, we used whole-genome sequencing method to find candidate mutations in 8 eat mutant strains previously believed to define 6 genes. We need more results to verify some of these candidates. However, these results, together with the identification eat‑17 as F01G12.6 and T24D11.1 (see Straud and Avery, this issue WBG) are likely to complete the molecular identification of all the eat genes.
Avery L. (1993) The genetics of feeding in Caenorhabditis elegans. Genetics 133, 897-917. 
Newman B, Gu X, Wintle R, Cescon D, Yazdanpanah M, Liu X, Peltekova V, van Oene M, Amos CI, Siminovitch KA. (2005) A risk haplotype in the Solute Carrier Family 22A4/22A5 gene cluster influences phenotypic expression of Crohn’s disease. Gastroenterology 128, 260–269.
Samuelson AV, Klimczak RR, Thompson DB, Carr CE, Ruvkun G. (2007) Identification of Caenorhabditis elegans genes regulating longevity using enhanced RNAi-sensitive strains. Cold Spring Harb Symp Quant Biol 72, 489–497.
Sarin S, Prabhu S, O’Meara MM, Pe’er I, Hobert O. (2008) Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat Methods 5, 865–867.
Seifried A, Schultz J, Gohla A. (2012) Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J, 549-571.
Wang GJ, Kang L, Kim JE, Maro GS, Xu XZS, Shen K. (2010) GRLD-1 regulates cell-wide abundance of glutamate receptor through post-transcriptional regulation. Nat Neurosci 13, 1489–1495.
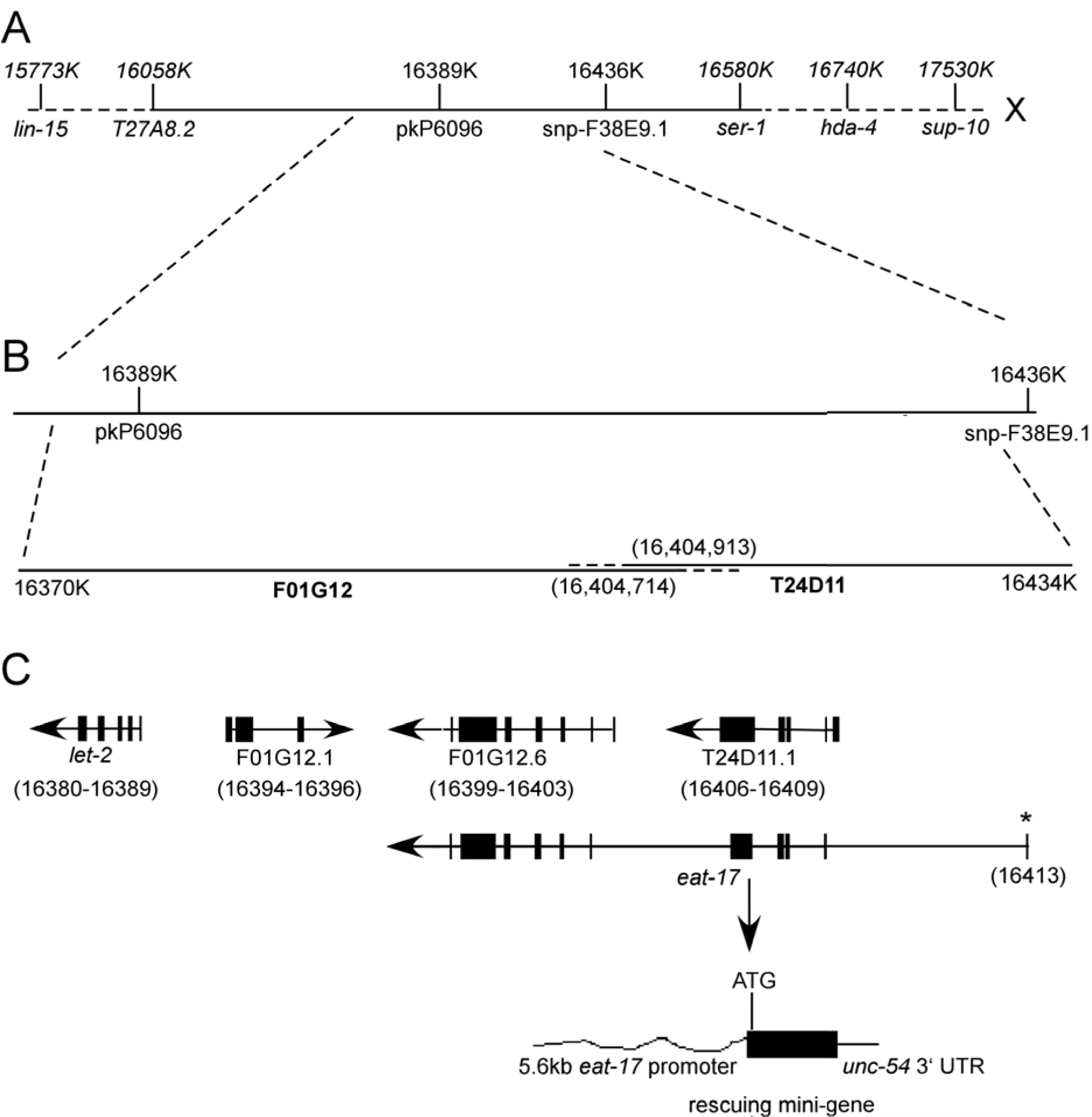
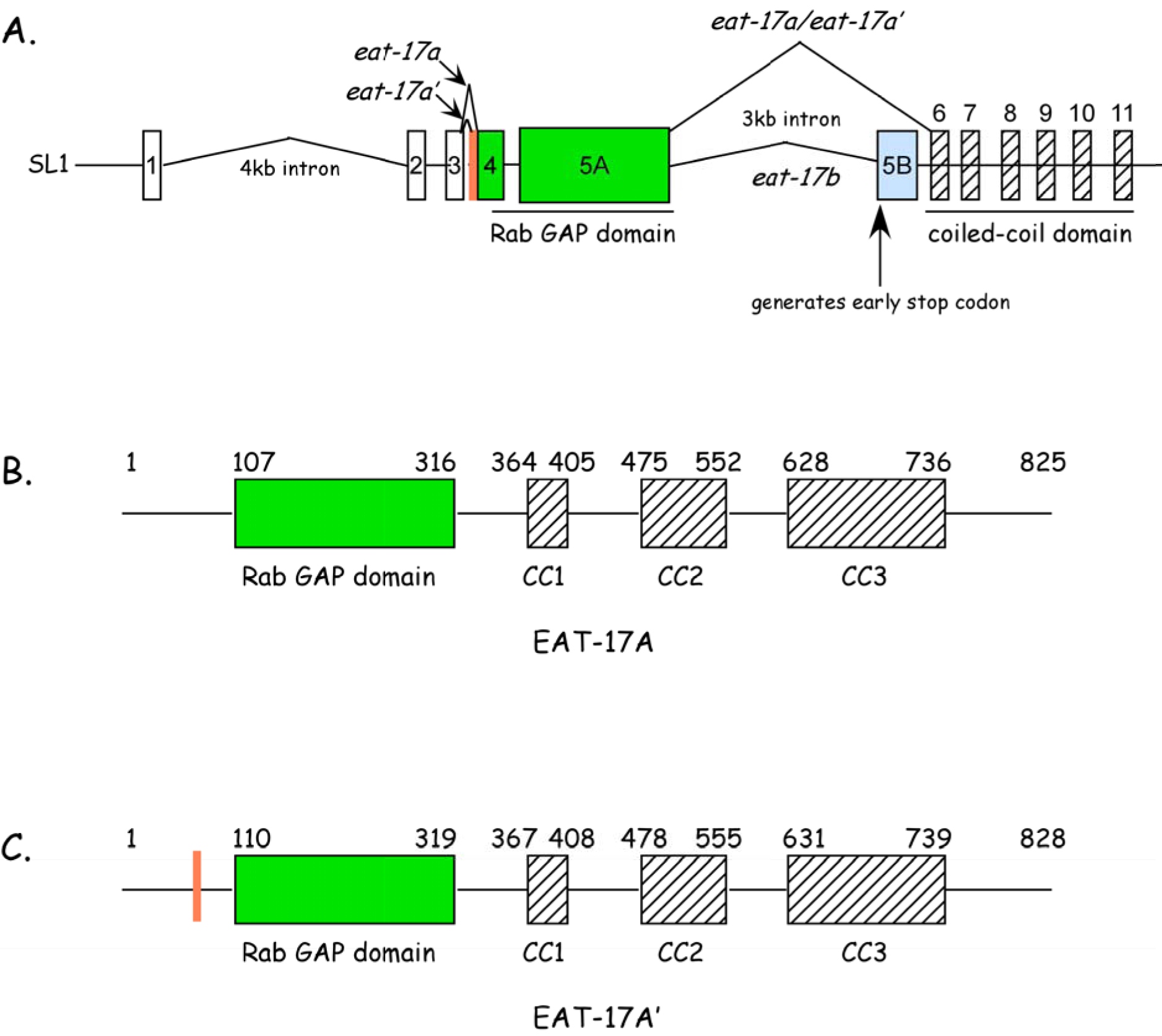
eat-17 was originally mapped to the right arm of Chromosome X, between lin-15 and sup-10. We used a three point mapping strategy combined with SNP mapping to narrow this down to a region between pkP6096 and snp-F38E9 (Figure 1). Co-injection of cosmids T24D11 and F01G12 was both necessary and sufficient to rescue feeding defects in a subset of transgenic progeny. One predicted gene in this region—with homology to the human Evi5 gene—became a strong candidate. Three key findings were used to show that this was the eat-17 gene. First, amplification and injection of the cDNA corresponding to this region was able to rescue the eat-17 mutant phenotype. Second, RNAi against this region in rrf-3 mutants resulted in phenocopy of the eat-17 mutant phenotype. Finally, we found a C to T base pair transition at nucleotide 607 in exon 5, creating a stop codon at amino acid 203, potentially rendering the protein non-functional.
Based on homology searches, we found that EAT-17 belongs to a family of Rab GAPs. By RT-PCR, we identified two transcripts, differing by 9 nucleotides. Two predicted proteins, 825aa and 828aa, respectively, are encoded by these two transcripts (Figure 2). Each contains a predicted RabGAP domain at the N terminus and a series of coiled coil domains at the C terminus. We used RNAi studies to identify RAB-6.2 as EAT-17’s interacting partner. RNAi of both eat-17 and rab-6.2 leads to defects in grinder formation. Though the role of these proteins in grinder formation is not clear, we suspect that both play an important role in trafficking collagens, chitin, or other components of the grinder to the apical surfaces of the terminal bulb muscles, particular during molts. Defects in other chitin based components, including the sieve, may explain defects in food trapping.
Of the 1090 somatic cells generated during development of C. elegans hermaphrodites, 131 undergo programmed cell death. Transcription factors that regulate cell-specific apoptosis in C. elegans have been associated with cancer (Reviewed in Potts et al., 2011). We aimed to identify novel transcription factors that regulate programmed cell death in C. elegans by performing an RNAi screen against transcription factors in a sensitized cell death defective background. A homolog of human leukemia associated AF10, zfp-1, was identified as a hit, as it was found to promote cell death in C. elegans. Our initial characterization of the programmed cell death defects of zfp-1 mutants is described here.
We performed an RNAi screen of 387 transcription factors and 263 chromatin remodeling factors using ced-3(n2427); Plin-11gfp worms to identify genes that regulate cell death in the ventral nerve cord. The Plin-11gfp reporter expresses GFP in the VC motor neurons of the ventral nerve cord, and provides a quick and reliable assay for identifying regulators of cell death (Reddien et al., 2001). In Plin-11gfp (otherwise genetically wild-type) worms (Figure 1A), four cells in the ventral nerve cord are clearly visible under a dissecting fluorescence microscope; two of the six VC neurons are obscured by fluorescence in vulval cells, which also express the reporter. In worms with mutations that result in a strong cell death defect, Plin-11gfp is expressed in five additional cells in the ventral nerve cord. These additional cells are the lineal equivalents of ventral neurons in the anterior and posterior ventral nerve cord, which undergo programmed cell death in wild type animals. In mutants with partial defects in programmed cell death, an intermediate number of additional cells survive and express Plin-11gfp. Worms defective in engulfment of cell corpses (ced-1(e1735)) or with a partial loss of function mutation in the caspase (ced-3(n2427)) have an intermediate number of additional cells that survive and express Plin-11gfp (Figure 1B, 1C). The sensitized ced-1(e1735) and ced-3(n2427) genetic backgrounds can enhance detection of weak cell death defects.
The strongest cell survival phenotype observed, both in the ced-3(n2427) background and in a secondary screen using ced-1(e1735), was in animals fed a zfp-1(RNAi) construct (data not shown). The zfp-1(ok554) deletion allele removes the leucine-zipper domain, rendering the protein non-functional (Cui et al., 2006). When zfp-1(ok554) is crossed into the Plin-11gfp reporter, there is a small, but significant, increase in the number of surviving ventral nerve cells (Figure 1D). The weak effect of zfp-1(ok554) in an otherwise wild type reporter background is enhanced in ced-1 and ced-3 mutants with partial defects in programmed cell death (Figure 1E, 1F). Other transcription factors, such as lin-35, can enhance a weak cell death defect (Reddien et al., 2007), and zfp-1 regulates a similar subset of genes (Grishok et al., 2008). It is possible that zfp-1 transcriptionally regulates cell death in a manner similar to that of lin-35. Thus, knockdown of zfp-1 might elicit a cell death phenotype as a result of its activity in transcriptional regulation.
Cui M, Kim EB, Han M. (2006) Diverse chromatin remodeling genes antagonize the Rb-involved SynMuv pathways in C. elegans. PLoS Genet 2, e74.
Grishok A, Hoersch S, Sharp PA. (2008) RNA interference and retinoblastoma-related genes are required for repression of endogenous siRNA targets in Caenorhabditis elegans. Proc Natl Acad Sci U S A 105, 20386–20391.
Potts M B, Cameron S. (2011) Cell lineage and cell death: Caenorhabditis elegans and cancer research. Nat Rev Cancer 11, 50–58.
Reddien PW, Andersen EC, Huang MC, Horvitz HR. (2007) DPL-1 DP, LIN-35 Rb and EFL-1 E2F act with the MCD-1 zinc-finger protein to promote programmed cell death in Caenorhabditis elegans. Genetics 175, 1719–1733.
Reddien PW, Cameron S, Horvitz HR. (2001) Phagocytosis promotes programmed cell death in C. elegans. Nature 412, 198–202.

{kind=link}