1. Laboratory of Aging and Neurodegenerative Disease, Center for Neurodegenerative Science, Van Andel Research Institute, Grand Rapids MI, USA
2. Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada
3. Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada
4. Division of Experimental Medicine, McGill University, Montreal, Quebec, Canada
5. Department of Genetics, Harvard Medical School, Boston MA, USA
2. Department of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada
3. Metabolic Disorders and Complications Program, and Brain Repair and Integrative Neuroscience Program, Research Institute of the McGill University Health Centre, Montreal, Quebec, Canada
4. Division of Experimental Medicine, McGill University, Montreal, Quebec, Canada
5. Department of Genetics, Harvard Medical School, Boston MA, USA
Correspondence to: Jeremy Van Raamsdonk (jeremy.vanraamsdonk@mcgill.ca)
In C. elegans, RNAi is a useful tool for decreasing the expression of a gene of interest. In order to properly interpret the results of an RNAi experiment it is important to quantify the level of gene knockdown typically using quantitative RT-PCR (qPCR). In some cases, qPCR primers will detect the RNA produced by the RNAi bacteria thereby leading to a false measurement of the mRNA levels of the gene of interest. To circumvent this problem, it is essential to design primer pairs in which at least one of the primers binds outside of the region targeted by the RNAi clone. As an example, we treated worms with an RNAi clone targeting the mitochondrial superoxide dismutase gene sod-2. When we compared gene expression to worms grown on EV RNAi using qPCR primers that bind within the sequence targeted by the RNAi clone, we detected no decrease in sod-2 mRNA level (Figure 1). However, when we examined sod-2 mRNA levels using primers that bind outside of the region targeted by the RNAi clone, we found that the sod-2 RNAi treatment did effectively reduce sod-2 mRNA levels (Figure 1). This illustrates the importance of designing primers such that they only amplify mRNA and not the RNA generated by the RNAi bacteria in order to properly measure the levels of knockdown.
Our standard approach to primer design for qPCR:
1. Go to www.wormbase.org
2. Search for gene of interest (e.g. sod-2)
3. Click on “Location”. This will show you if there are multiple transcripts. You may want to target a specific transcript or try to design primers that target all of them.
4. Click on “Sequences”. Under “Coding Sequence” all of the transcripts for the gene of interest will be displayed.
5. Copy the spliced RNA + UTR sequence to microsoft word (in order to keep a record of where your primers are targeting).
6. If you are designing qPCR primers to measure RNAi knockdown, identify the region of the transcript where the RNAi clone is targeting. At least one primer should be outside of this region. We find that often primers targeting the untranslated regions (UTR) bind outside of the RNAi targeted region.
7. We use Primer 3 to pick primers (bioinfo.ut.ee/primer3-0.4.0/). If possible, it is best to pick at least one primer that overlaps spans two different exons. By designing a primer that contains parts of two exons, this primer will not be able to bind to DNA, which has introns present. We normally aim for the amplified region to be 100-200 bp. In the word file, we normally indicate the primer locations and note the expected size of the amplified region.
8. As a final check, we run the primer pair through an in silico PCR website: http://genome.ucsc.edu/cgi-bin/hgPcr. Be sure to select C. elegans. Because the primers are specific for spliced RNA, they should not amplify any DNA sequences.
1. Go to www.wormbase.org
2. Search for gene of interest (e.g. sod-2)
3. Click on “Location”. This will show you if there are multiple transcripts. You may want to target a specific transcript or try to design primers that target all of them.
4. Click on “Sequences”. Under “Coding Sequence” all of the transcripts for the gene of interest will be displayed.
5. Copy the spliced RNA + UTR sequence to microsoft word (in order to keep a record of where your primers are targeting).
6. If you are designing qPCR primers to measure RNAi knockdown, identify the region of the transcript where the RNAi clone is targeting. At least one primer should be outside of this region. We find that often primers targeting the untranslated regions (UTR) bind outside of the RNAi targeted region.
7. We use Primer 3 to pick primers (bioinfo.ut.ee/primer3-0.4.0/). If possible, it is best to pick at least one primer that overlaps spans two different exons. By designing a primer that contains parts of two exons, this primer will not be able to bind to DNA, which has introns present. We normally aim for the amplified region to be 100-200 bp. In the word file, we normally indicate the primer locations and note the expected size of the amplified region.
8. As a final check, we run the primer pair through an in silico PCR website: http://genome.ucsc.edu/cgi-bin/hgPcr. Be sure to select C. elegans. Because the primers are specific for spliced RNA, they should not amplify any DNA sequences.
Figures
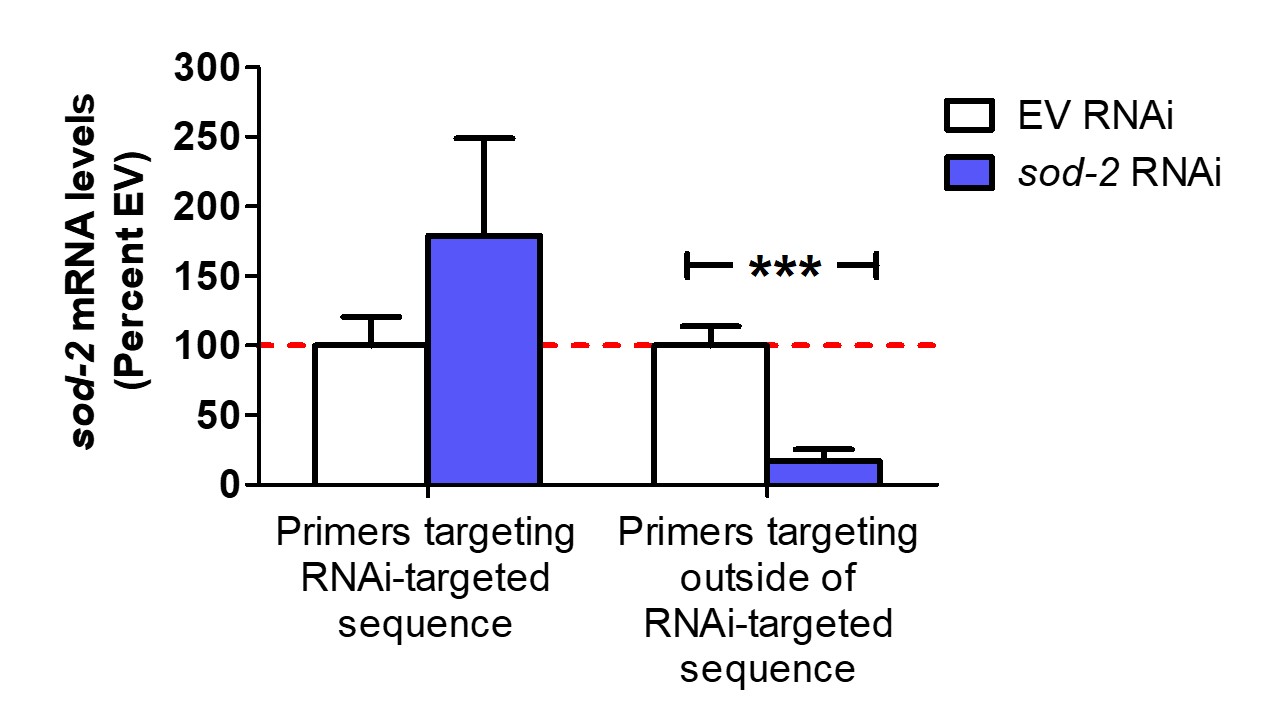
Figure 1: Using primers targeting outside of the RNAi-targeted sequence demonstrates that sod-2 RNAi effectively decreases sod-2 mRNA levels. Worms were grown on either empty vector (EV) or sod-2 RNAi bacteria beginning at the parental L4 stage. Gravid adults were transferred to a new plate, allowed to lay eggs for 24 hours and then removed. The resulting progeny were collected when they reached adulthood. RNA was isolated and converted to cDNA for quantification. Measuring sod-2 mRNA levels using primers that target the sequence targeted by the RNAi clone falsely shows little or no effect of sod-2 RNAi on sod-2 levels. In contrast, using primers that target outside of the RNAi-targeted sequence shows that sod-2 RNAi decrease sod-2 mRNA levels.