In our generation of a mutant in the gene T07C12.15 through CRISPR-Cas, we struggled at first with a PCR-only based approach. We then switched to using a phenotypic screen as described by Dan Dickinson and colleagues (Dickinson et al., 2013; website: http://wormcas9hr.weebly.com/). Through this screen, we were able to isolate mutants using either hygromycin- or the unc-119-based protocols. However, we made three interesting observations: 1) similar to other reports (Waaijers et al., 2013), we have noticed considerable embryonic and larval lethality in the F1, presumably owing to high level expression of the CRISPR-Cas9 endonuclease; 2) contrary to what has been suggested, we were able to relatively easily establish lines for the CRISPR-Cas9 transgenics; and 3) we noticed the few mutants that we have obtained (at a seemingly lower frequency than has been reported by others) might have arisen late in the growth/development of the injection plates.
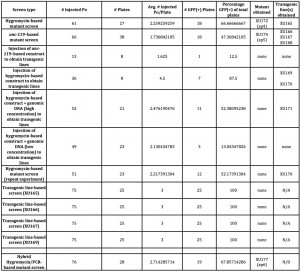
For these reasons, we wondered if it was possible that the mutant might arise through dsDNA breakage and transgene-based gene conversion later in the growth of the worms, as can occur in either endogenous transposon (Barrett et al., 2004)- or Mos1-based (Frokjaer-Jensen et al., 2010) mutagenesis. We set up experiments comparable to those done using the Dickinson protocol, except starting each plate with three transgenic animals derived from established transgenic lines. The results of these experiments are shown in Figure 1.
Unfortunately, we were not able to identify mutants through these lines, though our numbers (both of the number of animals and the rate with which we are obtaining the mutants) are low enough that it is worth repeating these experiments to be certain. The results with our lines are not unexpected and presumably due to silencing of the arrays. We have also tried coinjection of genomic DNA to overcome this obstacle (Figure 1), but such lines have proved to be nearly inviable (again presumably due to germline expression of the Cas9 endonuclease), and therefore seemingly unusable for this purpose.
We reasoned: if a mutant is obtained through processes occurring in the injected parent, then it should be possible to detect this mutant in the F1 generation, as has been observed by others (Friedland et al., 2013; Katic and Grosshans, 2013). We then realized that by combining the efficacy of a phenotypic hygromycin- or unc-119-based screen with a PCR-based approach, the whole process could be simplified, streamlined, and considerably accelerated.
Our modified workflow is shown in Figure 2, and the initial results of such an approach in the last row of Figure 1. By combining the hygromycin-based phenotypic approach with a PCR screen, we find that we are able to obtain a mutant in only four days from injection, and that the subsequent heat shock step to remove the transgenics may be unnecessary. With a hygromycin-based screen, after the second day on hygromycin, only the transgenics and putative mutants are completely healthy in appearance-and these latter two categories can be distinguished by the presence or absence of GFP or other markers. Candidate mutant animals can then be directly singled and tested by PCR, rather than waiting the additional 5-7 days to allow them to populate the plates and the additional time of subsequent heat shock followed by putative mutant selection. This hybrid PCR-phenotypic approach thus combines the “best of both worlds” and cuts the total time of mutant isolation from about 7-10 days, down to about four.
We thank Dan Dickinson for very helpful discussions over the course of these experiments.
Our modified workflow is shown in Figure 2, and the initial results of such an approach in the last row of Figure 1. By combining the hygromycin-based phenotypic approach with a PCR screen, we find that we are able to obtain a mutant in only four days from injection, and that the subsequent heat shock step to remove the transgenics may be unnecessary. With a hygromycin-based screen, after the second day on hygromycin, only the transgenics and putative mutants are completely healthy in appearance-and these latter two categories can be distinguished by the presence or absence of GFP or other markers. Candidate mutant animals can then be directly singled and tested by PCR, rather than waiting the additional 5-7 days to allow them to populate the plates and the additional time of subsequent heat shock followed by putative mutant selection. This hybrid PCR-phenotypic approach thus combines the “best of both worlds” and cuts the total time of mutant isolation from about 7-10 days, down to about four.
Figures

References
Barrett PL, Fleming JT, and Gobel V. (2004). Targeted gene alteration in Caenorhabditis elegans by gene conversion. Nat. Genet. 36, 11231-1237. 
Dickinson DJ, Ward JD, Reiner DJ, and Goldstein B. (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat. Methods 10, 1028-1034.
Friedland AE, Tzur YB, Esvelt KM, Colaiacovo MP, Church GM, and Calarco JA. (2013). Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 10, 741-743.
Frokjaer-Jensen C, et al. (2010). Targeted gene deletions in C. elegans using transposon excision. Nat. Methods 7, 451-453.
Katic I, and Grosshans H. (2013). Targeted heritable mutation and gene conversion by Cas9-CRISPR in Caenorhabditis elegans. Genetics 195, 1173-1176.
Waaijers S, Portegijs V, Kerver J, Lemmens BB, Tijsterman M, van den Heuvel S, and Boxem, M. (2013). CRISPR/Cas9-targeted mutagenesis in Caenorhabditis elegans. Genetics 195, 1187-1191.