
Oil pastel on paper by Beverley Dancy. Worm lettering and border by Andrea Choe.
View the full size version.
Oil pastel on paper by Beverley Dancy. Worm lettering and border by Andrea Choe.
View the full size version.
N1 still disgruntled over Brenner’s choice (Cambridge UK). Disproving the popular maxim that time heals all wounds, N1 made clear in a statement today that it is every bit as annoyed about not becoming the “chosen strain” as it was nearly 50 years ago. “Bristol had nothing on me then and it’s got nothing on me now,” said the visibly petulant Caenorhabditis elegans isolate obtained from Sidney Brenner’s backyard. “Mushroom compost? Give me a break,” added the derisive strain, referring to the humble origins of N2. According to N1, “It was totally political from the very beginning. These things always are. Anyway, it’s not like I really wanted to be prodded, mutagenized, starved, bleached and ultimately autoclaved by a bunch of sadistic scientists,” the transparently begrudging nematode added. “So the joke’s on you, N2. Hope you’ve enjoyed the last 50 years, because you’ve got a lot more of the same coming.”
Project dies peaceful death surrounded by loved ones (University of Davis, CA). Following a prolonged, uphill and often painful battle, NIH proposal GM052937 is finally at rest according to sources close to the proposal. Whereas the termination of the project came as something of a shock to its long-term collaborators, those closest to the revised R01 had expressed serious concerns in the months leading up to its death. “The signs were all there,” said one of the postdocs intimately familiar with the well-being of the project. “Insufficient preliminary data, a delay in publications, and a lack of connectivity to the aims. It was clear enough if you just looked.” The proposal’s brave but futile struggle was accompanied by emotions typical of end-of-project scenarios including a denial of experimental flaws, anger at reviewers and funding agencies, bargaining with editors, depression of summer salaries, and ultimately acceptance into a journal of significantly lower impact than was expected. Still, in all endings new beginnings may find their genesis. “It’s hard to know what lies beyond the mythical ‘great wall’,” stated one of the lead investigators after learning of the project’s demise. “Perhaps there’s nothing whatsoever. But maybe the Hindus have it right, and reincarnation awaits all things that pass.” As of press time, the project’s PIs were rumored to be heavily favoring the “theory of rebirth” in the form of an R21.
Worm theologians ponder meaning of ‘Pick of Fate’ (Petri dish). Ancient mythological narratives within the C. elegans community have long included fanciful tales of shining objects that descend from the sky, arbitrarily removing “chosen members” of the population. “Though it’s considered an ill omen to speak of, the Pick Gods must be appeased once in every generation,” proclaimed one worm elder, who spoke on condition of anonymity. Although reasons for the apparent sacrifices continue to elude the nematode clerics, it has been noted that virgin L4s appear to “please the giant gleaming spade” above all others. Some survivors of the choosing ceremony have reported approaching a bright light or flame. Still others claim that it is a form of alien abduction in which individuals are subjected to painful gonadal probing by a giant clear needle. Such survivors have described recovering consciousness on a completely different plate while smeared from nose to tail in a kind of viscous oily substance, possibly of an inter-dimensional or protoplasmic nature. Conspiracy theorists within the worm community have gone so far as to suggest that these individuals may have been “implanted” with a “seed of foreign origin”, although this theory remains controversial. Nevertheless, universal agreement behind the meaning of “Shovel Selection” is likely to remain unresolved. Stated one wizened post-gravid hermaphrodite, “We can only hope that a divine and loving intelligence is controlling the pick and that it does not merely represent the whims of some dispassionate cosmic manipulator.”
The C. elegans Development, Cell Biology & Gene Expression meeting is in Nara, Japan this July15th-19th, 2014. This meeting is in association with the 6th Asia-Pacific C. elegans Meeting.
Meeting Website: http://cedevap2014.sakura.ne.jp/
Abstract and registration opened March 9, 2014. Please pay attention to the deadlines.
Registration:
Early registration: May 9
Late registration: June 30
**On and after May 10, registration fee will be higher
Abstract Deadline: May 9
Financial Aid: We have received several grants to cover travel expenses of presenters (posters or talks). Please apply for this financial aid if needed.
Hotels: You must make your own hotel reservations by June 30th.
Hope to see you there!
Ahna Skop, Asako Sugimoto & Andrew Chisholm, Head Organizers
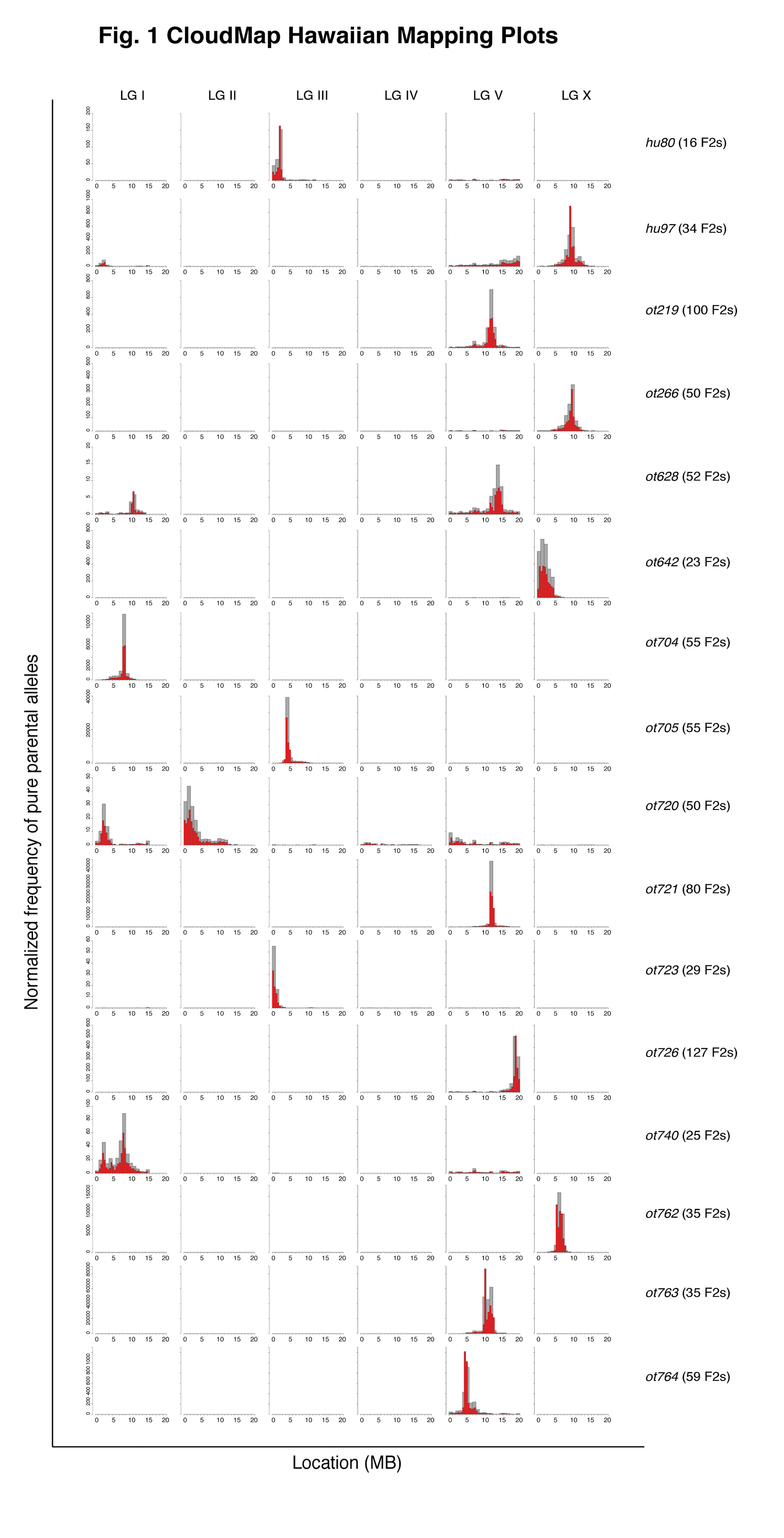
Our lab has previously developed a combination of single-step SNP mapping and whole genome sequencing (WGS) methods to pinpoint phenotype-causing sequence variants (Doitsidou et al., 2010). The mapping part of the single-step procedure is essential to separate the phenotype-causing, mutagen-induced sequence variant from background sequence variants. WGS data can be analyzed with CloudMap, a free, cloud-based software analysis tool that we recently developed (Minevich et al., 2012). We have previously provided proof-of-concept studies for the use of CloudMap, and we report here our experience with using CloudMap on a routine basis.
We have found over the last two years that the SNP/WGS strategy and ensuing CloudMap-based data analysis can reproducibly and accurately pinpoint phenotype-causing mutations to mapping regions as small as 0.5 Mb and that the method can work with very few recombinants. Fig. 1 shows the mapping plots from a selection of 16 EMS mutagenized strains from 8 different EMS-induced screens for loss of neuronal identity (thanks to many members of the Hobert lab for providing the mapping data). The tallest histograms indicate the regions containing the largest amount of normalized pure parental alleles following an outcross to the Hawaiian CB4856 mapping strain. For 14 of the causal variants in 13 strains (ot628 is a confirmed double mutant) we have either cloned the causal variant or have strong evidence for its identity and are in the process of confirming. For 8/14 of the causal variants, CloudMap correctly identified the 0.5 Mb where the causal variant resides. In the 6 cases where the 0.5 Mb bins did not correctly identify the causal variant, CloudMap was off by an average of 0.54 Mb.
The accuracy of CloudMap in identifying mapping intervals depends on strict adherence to the Hawaiian SNP mapping protocol and also on the number of F2 recombinants that are pooled for sequencing. In addition, Mendelian ratios should be calculated to identify whether the causal variant resides at one locus or many and if it is dominant or recessive. We also recommend thawing an ancestral Hawaiian strain once a year to minimize variants introduced by genetic drift and to maximize mapping accuracy.
In a number of cases where the Hawaiian mapping protocol was strictly followed, CloudMap was able to identify the correct 0.5 Mb mapping interval with very few recombinants. For example, both hu80 and hu97 were correctly mapped to a 0.5 Mb mapping interval with as few as 16 and 34 F2 recombinants respectively.
We consistently noted that the previously described incompatibility region between Bristol and Hawaian strains on the left arm of LG I (Seidel et al., 2008) is largely not visible relative to the peak of the causal variant due to the normalization procedure we use to compute the frequency of pure parental alleles. In cases where the causal variant is also on LG I (ot740, the taller peak contains the causal variant), CloudMap was able to separate the incompatibility peak from the causal variant peak.
Transgenes contained in the strain background and required to observe a mutant phenotype can generate a mapping signal if the transgene is re-homozygosed in the F2 generation. We find that this mapping signal is virtually eliminated if as few as 50% of the picked F2 recombinants are heterozygous for the transgene.
CloudMap is available via the Galaxy web platform, requires no installation when run on the cloud, and can also be run locally or via the Amazon Elastic Compute Cloud (EC2) service (http://usegalaxy.org/cloudmap and http://hobertlab.org/cloudmap).
Doitsidou M, Poole RJ, Sarin S, Bigelow H, and Hobert O. (2010). C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 5, e15435. 
Minevich G, Park DS, Blankenberg D, Poole RJ, and Hobert O. (2012). CloudMap: A cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269.
Seidel HS, Rockman MV, and Kruglyak L. (2008). Widespread genetic incompatibility in C. elegans maintained by balancing selection. Science 319, 589–594.
Mutation mapping by whole-genome sequencing (WGS) is becoming increasingly popular with C. elegans researchers. Pioneering studies from several worm labs have laid the foundations for this trend by providing proofs of concept (Sarin et al., 2008; Flibotte et al., 2010), working out experimental strategies (Doitsidou et al., 2010, Zuryn et al., 2010), and contributing tools and workflows for the analysis of mapping-by-sequencing data (Minevich et al., 2012).
Yet even though many good bioinformatic tools for WGS data analysis exist, almost all of them seem daunting to biologists. The CloudMap collection of workflows and tools for Galaxy is a notable exception here, but it requires the upload of huge experimental datasets to the main Galaxy server (http://usegalaxy.org), which is time-consuming and not well-suited for maintaining an archive of data from several experiments. Hence, most people in the field still do not consider analyzing WGS data themselves, and the resulting need to establish collaborations prevents a more rapid spread of mapping-by-sequencing approaches in the community.
We have recently developed MiModD, a comprehensive software package for the identification and annotation of mutations in the genomes of model organisms from whole-genome sequencing data, with the specific goal to enable biologists/geneticists with limited bioinformatical knowledge to analyze genome-wide sequencing data locally on (relatively) standard desktop computers. The current beta version of MiModD enables short-reads alignment from several input formats, variant calling, post-processing (i.e., filtering and annotating lists of variants), and the generation of summary reports with hyperlinks to biological databases including Wormbase. It also supports various mapping-by-sequencing approaches by producing CloudMap-ready output, i.e., variant lists produced by MiModD can be used directly with the CloudMap EMS Variant Density, Variant Discovery Mapping, and Hawaiian Variant Mapping tools. By default, the package is controlled through a command line interface, but can be fully integrated, with a single command, into a local installation of Galaxy to obtain a beginner-friendly graphical user interface and a lab-internal WGS analysis server.
In principle, MiModD can be used to analyze WGS data from any organism, but its memory requirements are dependent on genome size. To analyze C. elegans data we recommend a system with 16GB of memory (though 8GB will do for testing). The package runs on Linux and Mac OS systems without any further strict hardware requirements. Obviously, performance will depend on the exact configuration of the system MiModD is running on, but on our development machine – a desktop PC equipped with a 3rd generation Intel Core i7 processor, 16 GB RAM and an Ubuntu 12.04 operating system – the complete analysis of a 30x covered worm genome finishes in less than 1 hour, which should be good enough for the occasional user.
MiModD is free, open-source and actively developed, and labs interested in WGS analysis are encouraged to download and test the software. We offer assistance with installing and using the package to users of the beta version and are looking forward to receiving your feedback. Further information is available at www.celegans.de/mimodd.
Sarin S, Prabhu S, O’Meara MM, Pe’er I, and Hobert O. (2008). Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods 5, 865-867.
Flibotte S, Edgley ML, Chaudhry I, Taylor J, Neil SE, Rogula A, Zapf R, Hirst M, Butterfield Y, Jones SJ, et al. (2010). Whole-genome profiling of mutagenesis in Caenorhabditis elegans. Genetics 185, 431-441.
Doitsidou M, Poole RJ, Sarin S, Bigelow H, and Hobert O. (2010). C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One 5, e15435.
Zuryn S, Le Gras S, Jamet K, and Jarriault S. (2010). A strategy for direct mapping and identification of mutations by whole-genome sequencing. Genetics 186, 427-430.
Minevich G, Park DS, Blankenberg D, Poole RJ, and Hobert O. (2012). CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249-1269.