
View the full size version.
WormBase private instances via Amazon’s Cloud infrastructure
1Hi-Line Informatics, LLC. Livingston, MT, 2Ontario Institute for Cancer Research, Toronto ON, Canada
Correspondence to: Todd Harris (todd@wormbase.org)
At WormBase, we are often asked if it is possible to install and run a local version of the website. Although certainly possible and very well documented, it’s not recommended for three reasons. 1) The size of the databases require substantial download time that must be repeated on a monthly basis to maintain an up-to-date resource; 2) The site is complex and requires significant time to install and configure; moreover, it is constantly evolving. You’ll need to commit time to keeping your site up-to-date; 3) Finally, the site requires a substantive compute environment along with concomitant system administration acumen required to keep everything up and running smoothly. If you still aren’t dissuaded, please see the installation notes on the WormBase Wiki.
But now — through the magic of cloud computing — you can have your own WormBase up-and-running in a few minutes.
Required Steps
- Establish an account on Amazon Web Services
- Find and launch the WormBase Amazon Machine Image (AMI) of the version of your choice.
- Connect to the newly launched server instance using your web browser.
- Stop the instance when done to avoid incurring further charges.
- Repeat steps 2-4 when a new version of WormBase is released.
Intended Audience
- Individual researchers or labs
- Entire departments
- Private research entities
Necessary Skills
- For launching an instance: none beyond using a web browser
- For more complicated data mining: command line expertise
Suggested Uses
- Access your own WormBase: speedy and it’s private
- Data mining: all databases preconfigured; includes common tools like BioPerl
- Development: build new features using the WormBase web platform
Caveats
- This is NOT a free service. Read the pricing details carefully.
- Although we will release new AMIs for each release, your instances will not receive bug updates.
For more information and a detailed walkthrough of the process, please see An introduction to cloud computing for biologists.
Keeping up with the changes
1Centre d’Immunologie de Marseille-Luminy, INSERM U631, Marseille, France
Correspondence to: Jonathan Ewbank (ewbank@ciml.univ-mrs.fr)
In common with many labs, in the past we routinely used DAVID to look for over-representation of functional classes in lists of genes. Unfortunately, the data in DAVID hasn’t been up-dated since September 2009. As an alternative, there’s a stand-alone version of DAVID called EASE. This allow labs to use their own lists. A post to the forum last year asking whether anyone maintained up-to-date EASE lists went unanswered, so we decided to make our own. That’s when we realized that there are three challenges. One is simply having a user-friendly interface to enter any new gene list, with the appropriate citation information. The second is converting any gene list culled from the literature into a coherent form. This is made more difficult by the fact that one still finds gene lists in published papers that (i) contain an apparently random mix of different types of gene identifiers and (ii) don’t say which WormBase WS version was used. The third is that with changes in predicted gene structure (genes being split, killed, fused, etc), lists become increasingly inaccurate with time.
We therefore made a first tool, the WormBase Converter. It can automatically detect different IDs (Transcript Name, Gene Name, WormBase ID, etc.) and convert them into the type of ID you need. It also allows the conversion between WormBase releases. You can convert a Gene ID from a specific release (e.g. WS170) into another release (e.g. WS220 for a conversion, or WS160 for a reverse-conversion).
As WormBase Converter was originally made for use with EASE, we didn’t design it to give a correspondence table, but are currently working on adding this as an option, so that there will be the possibility of having an output table listing in one column the input genes and in an adjacent column the list of genes in the required output format. There are stand-alone WormBase Converter versions for all common platforms (Unix, MacOS, Windows) available on Sourceforge.org. As keeping it up to date does take some effort, due to the need to correct the occasional Wormbase annotation anomaly, we also offer a client version that sends requests to the WormBase Converter server that we maintain at the CIML. We’d be delighted if anyone else would like to host this service!
We also made EASE Manager. This facilitates the entry and management of gene lists into EASE, but more importantly, interfaces with the WormBase Converter so that the genes that make up any list are kept up to date. Installation instructions and full documentation can be obtained from our website. We welcome feedback on these tools, which we also described in a paper earlier this year (Engelmann et al., 2011).
Figures
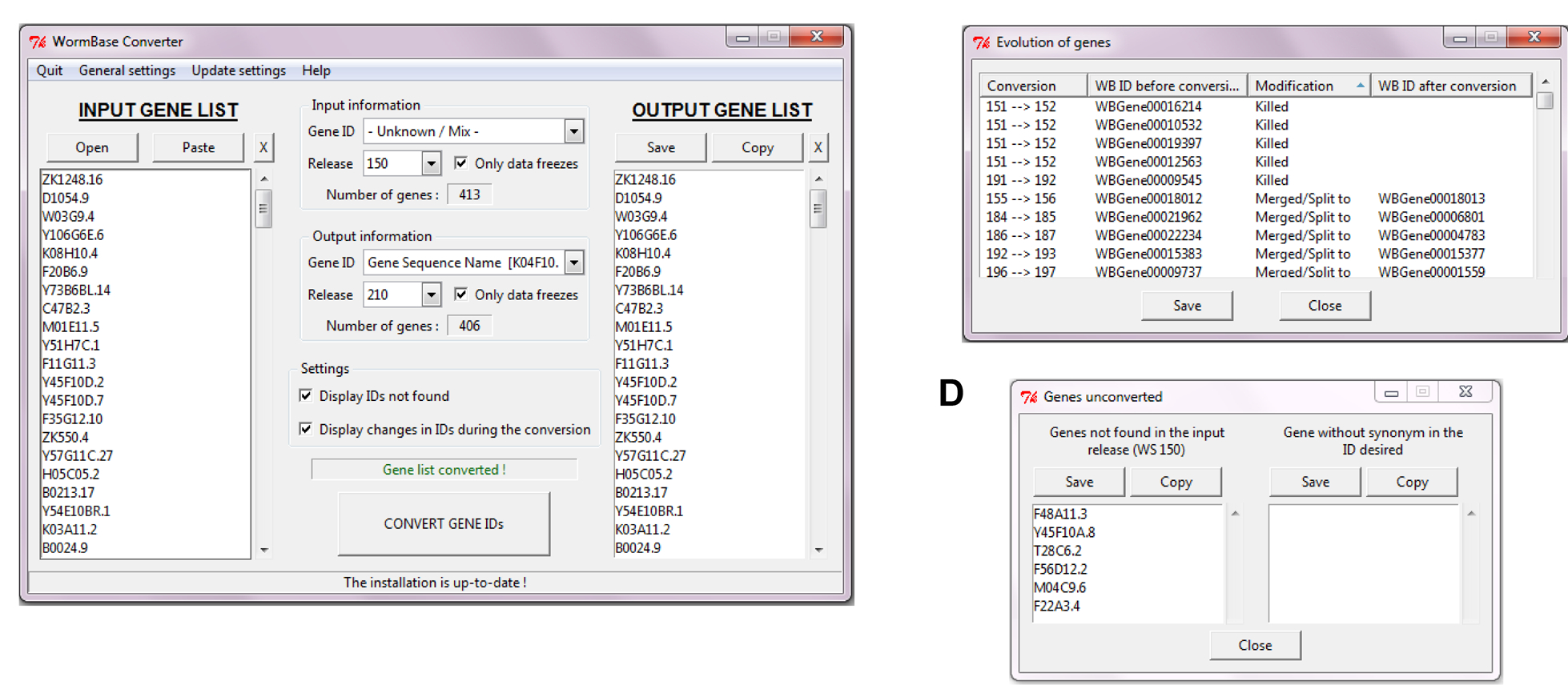
Figure 1: Figure taken from Engelmann et al., 2011
References
Engelmann I, Griffon A, Tichit L, Montañana-Sanchis F, Wang G, Reinke V, Waterston RH, Hillier LW and Ewbank JJ. (2011). A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS One 6, e19055. 
HOT operons!
WormBase Group, Wellcome Trust Sanger Institute, Hinxton, Cambridge, UK
Correspondence to: Gary Williams (gw3@sanger.ac.uk)
The modENCODE ‘Integrative Analysis’ paper (Gerstein et al., 2010) described 304 unusual transcription factor binding site regions which they named ‘Highly Occupied Target (HOT) regions’. These short (~400 bp) regions bind to 15 or more transcription factors and are not enriched in transcription factor motifs when compared to the normal specific binding sites. The HOT regions are associated with genes which are highly expressed and which are more likely to be essential compared to other genes. The HOT regions are included in WormBase as the features ‘WBsf216780’ to ‘WBsf217083’, inclusive.
The HOT regions tend to be associated with the 5′ region of operons. This association was not commented upon by the authors of the modENCODE paper (Gerstein et al., 2010).
82 of the HOT regions are located within 2 Kb of the 5′ end of operons. 20 of these are within 2 Kb of two operons flanking them on both strands. Operons sometimes have genes at either end which have not been included in them for lack of evidence, or have genes added to them which should not be included. If the distance from the start of the operon is extended to 10 Kb to allow for the uncertainty in the start position of an operon, then the number of HOT regions located near the 5′ end of an operon increases to 143, and the number of these with two flanking operons increases to 40.
Assuming the null hypothesis that HOT regions should associate with the genes at the 5′ end of operons as frequently as with any other coding gene, the binomial distribution gives a significant result for the observed association (p-value < 2.2e-16).
HOT regions may be useful for locating operons which have not been curated so far. Several such potential operons have been seen during a cursory inspection of those HOT regions which are not near a known operon. HOT regions will not however be used by the WormBase curators as evidence for the existence of a nearby operon because HOT regions are just as likely to control genes with a property like being constitutively expressed (which could preferentially include operon genes) as they are to be specific markers for the 5′ end of operons.
Figures
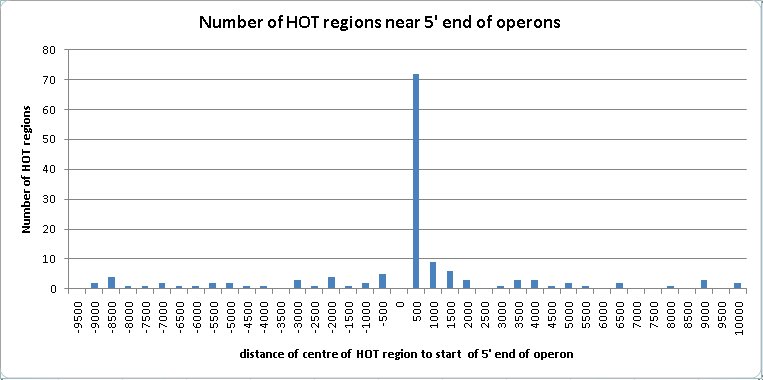
Figure 1: The clustering of the centre point of HOT regions near the 5′ end of operons peaks in the first 500 bases upstream of the operons.
NucleiTracker4D: A toolbox for semi-automated analysis of embryonic lineages and morphogenetic movements
1Division of Biological Sciences, University of California, San Diego CA, 2Department of Electrical and Computer Engineering, University of California, San Diego CA
Correspondence to: Claudiu A. Giurumescu (cgiurumescu@ucsd.edu)
Automated tracking of fluorescently labeled nuclei in 4D data sets has been used for several years for cell lineage tracing (Bao et al., 2006) and mapping gene expression patterns in C. elegans (Murray et al., 2006). Fully automated tracking of nuclei during late morphogenetic movements (i.e. above 350 cells) has remained challenging owing to the increasing density of nuclear packing in the later embryo.
We have developed a new tool for tracking labeled nuclei that avoids the increasing error rates associated with fully automated lineaging. Our approach is semi-automated: starting with a curated map of all nuclei in a 4D frame at any given time point, our software conducts local searches for nuclei at the next time point. Any nucleus that appears to have moved less than a threshold distance (0.75 of its radius) is assumed to be the same nucleus as in that position at time t. Nuclei that have moved greater than the threshold distance, or new nuclei arising by division, are flagged to be curated manually.
Using this approach we can trace all nuclei in individual embryos up to the 1.5-fold stage (670 nuclei; Figure 1), after which muscle movements make tracking difficult. It takes approximately 8 hours to track nuclei up to the 350-cell stage, and 80-100 hours to the 1.5-fold stage. Our software generates visualizations of the 4D data sets that can plot nuclear velocities and trajectories over chosen intervals. Our software has been used on 4D movies of histone-GFP (zuIs178) labeled nuclei acquired on a Zeiss LSM510 confocal and saved in the Zeiss .lsm format. We have been able to track nuclei in other confocal 4D data sets, the limiting factor being blurring of nuclear GFP signal at the top of a z-stack as nuclear density increases. In principle our approach can be applied to any sample in which many objects are being tracked over time. Our code is available at http://sourceforge.net/projects/nucleitracker4d/. NucleiTracker4D v2.0 requires a computer with at least 4 Gb of RAM as well as Matlab and its image processing toolbox. We welcome feedback from the community.
Figures

{kind=link}
References
Bao Z, Murray JI, Boyle T, Ooi SL, Sandel MJ and Waterston RH. (2006). Automated cell lineage tracing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 103, 2707-2712.
Murray JI, Bao Z, Boyle TJ and Waterston RH. (2006). The lineaging of fluorescently-labeled Caenorhabditis elegans embryos with StarryNite and AceTree. Nat. Protoc. 1, 1468-1476.