Worm Breeder's Gazette 8(3): 21
These abstracts should not be cited in bibliographies. Material contained herein should be treated as personal communication and should be cited as such only with the consent of the author.
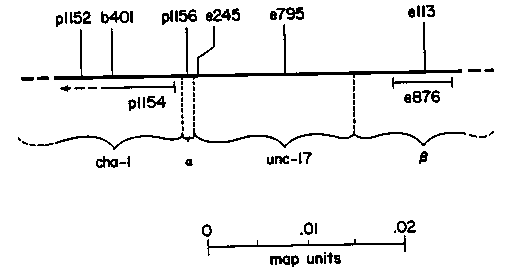
The gene cha-1 IV is the structural gene for choline acetyltransferase (ChAT), and maps very close to unc-17. unc-17 strains have the same set of phenotypes as cha-1 strains (including resistance to cholinesterase inhibitors), except that they have apparently normal ChAT levels. Complementation analysis revealed three (or four) classes of alleles (Rand and Russell, Genetics 106:227) . There were cha-1 alleles and unc-17 alleles, each forming a discrete complementation group, and in addition, a set of three anomalous alleles, which appeared to be members of both complemention groups. Among the anomalous alleles, there were two subgroups: the alpha class, which at the moment contains only the p1156 allele and is ChAT-deficient, and the class, consisting of e113 and e876, which when homozygous lead to qualitatively different growth and behavior, and near-normal ChAT levels. In order to explain the complementation pattern and the similarity of phenotypes, we proposed a two-domain model of ChAT function at the 1983 Cold Spring Harbor Meeting. That model suggests that the ChAT polypeptide has two functionally independent and structurally discrete domains, one of which (the one disrupted by cha-1 mutations) is responsible for the enzyme's catalytic function, and the other (disrupted in unc-17 mutations) involved in some essential function such as proper subcellular localization of the enzyme. The model also requires that the ChAT molecule function in vivo as a homodimer, and suggests that the alleles lead to defects in dimerization as well as 'localization'. I have recently been constructing a fine-structure map of the region. Heteroallelic animals were generated carrying pairs of noncomplementing cha-1 and/or unc-17 alleles; in addition, one chromosome carried the left-flanking marker lin-1 and the other chromosome had the right-flanking marker dpy-13. For each pair of alleles tested, about 300-500 such heteroallelic animals were generated, and about 50,000-120,000 self-progeny of such animals were scored for normal movement. In most cases, some wild-type animals were identified, and progeny tests of these individuals led to unambiguous left-right ordering. By checking for exchange of the flanking markers, I was able to confirm that such individuals had arisen by a recombination event between the two alleles tested, and not by apparent gene conversion. The current version of the map is shown below. Alleles which have been ordered unambiguously with respect to each other are shown above the line; the possible extents of alleles not yet fully mapped are indicated below the line. The distances between alleles indicate the approximate recombination frequency. The following observations can be made: 1. The map so far is entirely self-consistent; i.e., in all cases, the left-right ordering relationship is transitive and the map distances are approximately additive. 2. The different types of alleles are not interspersed, but seem to be in clusters - all of the cha-1 alleles are to the left of the unc- 17 alleles (although admittedly there are not yet many of the unc-17's on the map). 3. The two kinds of anomalous alleles map differently. The single alpha allele p1156 maps between the cha-1 and the unc-17 alleles, while both of the alleles, e113 and e876, map to the right of the unc-17 alleles. 4. The size of the region, as determined by recombination frequency, is rather large - about 0.03 map units. This is considerably larger than recombination frequencies observed between alleles within the unc- 13 or unc-15 genes, for instance. This may merely be a side-effect of being outside the main gene cluster on Linkage Group IV, or it may reflect large and/or many introns in the gene (the ChAT protein has an apparent molecular weight of 71,000 daltons). These results are entirely consistent with (but by no means prove) our two domain model. Thus, each of the domains, as inferred from the complementation analysis, would be a discrete bead on the string of the map. Nevertheless, examination of the map suggests other possibilities. We have thus begun to entertain an alternative model, involving a single message that can be spliced in different ways. [See Figure 1]