Worm Breeder's Gazette 16(1): 28 (October 1, 1999)
These abstracts should not be cited in bibliographies. Material contained herein should be treated as personal communication and should be cited as such only with the consent of the author.
Division of Molecular Biology, The Netherlands Cancer Institute, Center for Biomedical Genetics, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands
It is quite clear that single nucleotide polymorphisms (SNPs) are a going to be a useful source of genetic mapping information in C. elegans and other organisms with a sequenced genome. However, there are several limitations to current approaches to SNP detection and mapping. First, we have found that it is necessary to confirm the presence of each SNP experimentally; potential SNPs are occasionally due to either sequencing or cloning artefacts. Second, most reported SNPs have been derived from a single wild isolate (CB4856). Although this strain appears to be a good choice in terms of providing a relatively even spread of polymorphisms in the genome (Koch et al, in preparation), we have found that some phenotypes (i.e. chemotaxis) cannot be mapped using this strain, because CB4856 occasionally fails to phenocopy N2. Finally, although sequencing is, in principle, easy and reliable, it can be expensive, especially if hundreds of recombinants, many of which are heterozygous for the SNP, need to be genotyped.
To address some of these concerns we have undertaken a project to identify and verify a large number of SNPs that can be detected by RFLP as a consequence of the modification of a restriction enzyme recognition site. These "snip-SNPs" are then easily visualised by simply running an agarose gel after digestion of a PCR product with the appropriate enzyme. Several other advantages are: 1) the PCR product does not need to be purified (direct amplification and cutting of single-worm lysates in one PCR tube works well for most tested enzymes), 2) the cost and speed per sample is much lower than sequencing (important when hundreds of genotypes per cross are analysed), 3) heterozygous genomes can be reliably identified and 4) as with most SNPs (but unlike visibles) most snip-SNPs are phenotypically silent. The use of snip-SNPs does not preclude the use of other SNPs. We have found that these snip-SNPs are particularly useful to establish linkage, and can provide a relatively small interval within which to check for regular SNPs. Many crossovers can be mapped well without sequencing.
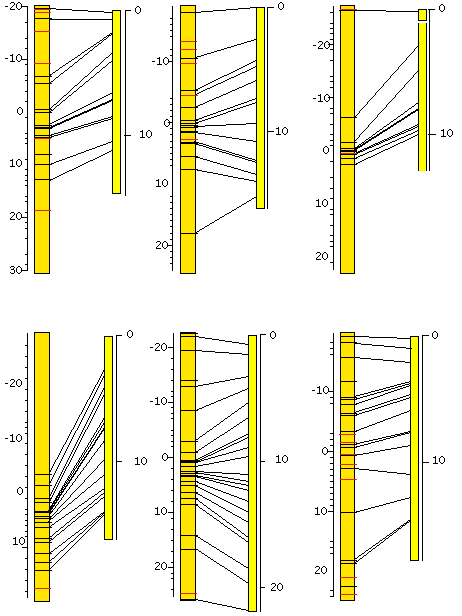
To date, we have verified more than 100 snip-SNPs distributed over the genome. Further searching will be restricted to those areas which as yet are poorly covered. Once the set is complete we will deposit these confirmed polymorphisms–along with the information required to detect them–in a publicly available SNP database. A subset of those are shown in Figure 1. Furthermore, to address the issue of phenotypic variance between CB4856 and N2, we have selected 10 F2 cross progeny from a cross between these two strains. These 10 CB4856/N2 hybrid strains have been selfed through 13 generations and are currently being genotyped for a subset of the available snip SNPs. These then, can be phenotyped for a given trait, and the region of interest to the mapper can be checked for the relevant snip-SNPs. This should broaden the variety of phenotypes which can be mapped using CB4856.
Figure 1: The positions of confirmed snip-SNPs on the genetic (left) and physical (right) maps. Genetic map positions were interpolated.