Worm Breeder's Gazette 13(4): 72 (October 1, 1994)
These abstracts should not be cited in bibliographies. Material contained herein should be treated as personal communication and should be cited as such only with the consent of the author.
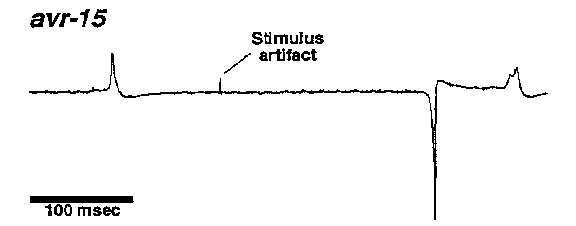
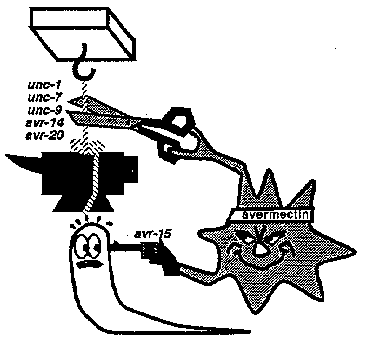
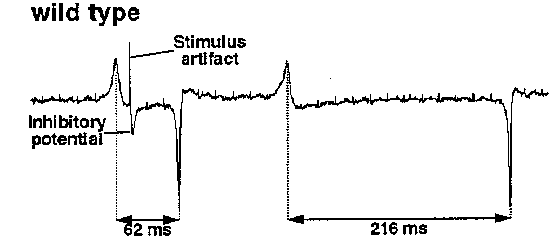
[1]Department of Biochemistry, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75235-9038. [2]Department of Biological Computation, AT&T Bell Labs, 600 Mountain Ave, Murray Hill, NJ 07974. [3]Department of Biochemistry, Cornell University, 217 Biotechnology Building, Ithaca, NY 14853-2703. M3 is a fast inhibitory motor neuron, producing negative postsynaptic voltage changes that can trigger pharyngeal muscle relaxation (Raizen and Avery, Neuron 12: 483). We now show that M3's neurotransmitter is glutamate, and suggest that it acts via the glutamate- gated Cl- channel cloned by Cully et al (1993 Worm Meeting 177) as an avermectin receptor. AVR-15 IS NECESSARY FOR M3 TRANSMISSION Because M3 transmission is fast and inhibitory, we thought it might be mediated by a ligand-gated Cl- channel. unc-25 and mutants, which lack GABA-A transmission, have normal M3 transmission, excluding the GABA-A receptor. We therefore focused on the avermectin-sensitive glutamate-gated Cl- channel. We tested a collection of avermectin- resistant mutants isolated by Carl Johnson, and found that avr-15 mutants had no detectable M3 transmission. In addition, by direct screening for mutants with abnormal feeding and pharyngeal electrophysiology, we isolated a mutation ad1051 that lacks M3 inhibitory potentials and fails to complement Carl's avr-15 alleles. GLUTAMATE PULSES MIMIC THE ACTION OF M3 ON PHARYNGEAL MUSCLE If avr-15 controls the avermectin-sensitive glutamate receptor and this receptor mediates M3 transmission, wild-type but not avr-15 pharyngeal muscle should be inhibited by glutamate. We first tried to test this prediction by dissecting pharynxes into a bath containing glutamate. 0.5 mM glutamate inhibited wild-type and avr-15 pharynxes equally. There were two general interpretations of this result: (1) Our hypothesis was wrong: M3 does not talk via glutamate. (2) The inhibition of pumping by 4 min exposure to 0.5 mM glutamate was a non-specific effect unrelated to M3 transmission. To distinguish these possibilities, we needed a more physiological way to apply glutamate. We dissected pharynxes into a solution containing caged glutamate, which was then photolyzed with a flash of UV light, releasing a brief pulse of glutamate. The pulse of glutamate mimicked M3 in two ways: (1) It caused a brief inhibitory potential. (2) It decreased overall pump duration. (Compare the pump during which the pulse occurred to the one that follows.) The response depended on light and caged glutamate. Both effects could be repeated for as long as the pharynx stayed alive, and worked for four pharynxes. [Figure 1] We also reproduced the effect on duration by a different method of applying glutamate pulses: ionophoretic ejection from a pipet. This experiment has been done on more than ten pharynxes. GLUTAMATE PULSES HAVE NO EFFECT ON AVR-15 MUTANT MUSCLE. We have only done the caged glutamate experiment on one avr-15 pharynx so far. As shown in the figure, it did not respond. We have ionophoresed glutamate onto several avr-15 pharynxes, and in our preliminary experiments have seen no response. [Figure 2] M3 NEURONS STAIN WITH ANTIBODIES AGAINST GLUTAMATE On a visit to Boston, Anne Hart and Josh Kaplan showed one of us worms stained with antibodies against glutamate. The cell bodies of the M3 neurons were clearly stained. A few as yet unidentified neurons in the terminal bulb also stained. GLUTAMATE PULSES HAVE THE NORMAL EFFECT ON EAT-4 MUSCLE. avr-15 is the second gene we know necessary for M3 transmission. Strong eat-4 mutants also lack M3 transmission. eat-4 mutant pharyngeal muscle responds normally to ionophoretic glutamate pulses. This result suggests that eat-4 acts in M3 -- that M3 fails to release glutamate. The eat-4 phenotype is consistent with failure of all glutamatergic transmission. In addition to their pharyngeal defect, which is similar to that of avr-15 or M3- worms but stronger, eat-4 mutants are defective in response to nose touch (A Hart and J Kaplan, personal communication), a phenotype they share with not-3 mutants, which lack an excitatory glutamate receptor (V Maricq, A Hart, J Kaplan, and C Bargmann, personal communication). eat-4 mutant behavior does not suggest defects in acetylcholine, serotonin, or GABA transmission. eat-4 may correspond to ZK512.6, which has sequence similarity to membrane transporters (Lee and Avery, WBG 12(5): 65; Ni et al, PNAS 91: 5607). Our current favorite guess is that eat-4 encodes the transporter that concentrates glutamate in synaptic vesicles. SPECULATION ON THE GENETICS OF AVERMECTIN SENSITIVITY Carl Johnson's genetic studies suggest that in C elegans avermectin has two distinct targets: one (presumably the pharyngeal glutamate-gated Cl- channel) controlled by avr-15, and another controlled by unc-1, 7, 9, and avr-14, and 20. Since the defect produced by loss of M3 transmission is mild, we imagine that the importance of the avr-15 target might vary from species to species, perhaps explaining on the one hand why only a single mutation is necessary for resistance in C briggsae (C Johnson, 1993 Worm Meeting 224), and on the other hand the extremely high pharyngeal sensitivity to avermectin in Haemonchus contortus (Geary et al, Exp Parasitol 77:88). If the pharyngeal glutamate-gated Cl- channel is phylogenetically variable, the target controlled by the five genes might be more important for broad-spectrum anthelminthic activity. [Figure 3] ACKNOWLEDGMENTS We thank Carl Johnson for giving us his avermectin-resistant mutants, and Anne Hart, Josh Kaplan, and Carl for unpublished information and ideas.