Worm Breeder's Gazette 13(1): 62 (October 1, 1993)
These abstracts should not be cited in bibliographies. Material contained herein should be treated as personal communication and should be cited as such only with the consent of the author.
The site at which 3' ends of mRNAs are formed by cleavage and polyadenylation has been well characterized in vertebrates. The cleavage occurs about 15 - 25 nucleotides downstream of the sequence, AAUAAA, and virtually no variation in this sequence is tolerated. In contrast, it has not been possible to identify an equivalent sequence in yeast. To initiate a molecular analysis of mRNA 3' end formation in worms, we have analyzed a random C. elegans cDNA library whose 3' sequences have been determined. This library includes more than 1500 sequenced clones, and our analysis indicates that fewer than 20% are duplicated. So cDNAs from about 1300 different genes have been included in the study.
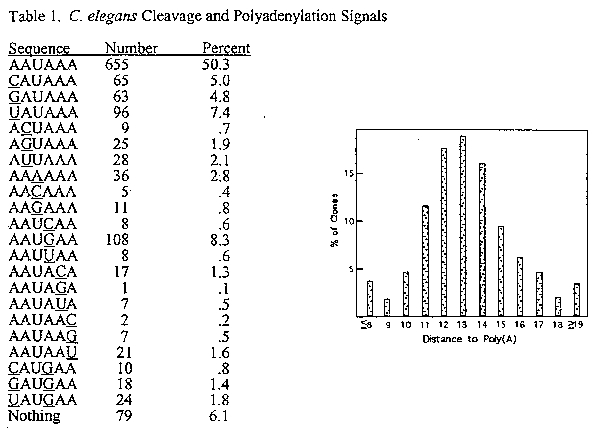
As soon as sequences from a few cDNA and genomic clones from C. elegans were known, it was clear that C. elegans does use the AAUAAA signal, but that variation is tolerated. Thus, to begin our analysis, we searched for perfect matches to AAUAAA, and found them an appropriate distance (see below) from the poly(A) in about half of the clones. We then searched for all possible one-base mismatches, and found them in the right place in most of the remaining clones. Finally, we looked at all of the clones that had no close match, and found three different 2-base mismatch sequences that appear to serve this function in a significant number of instances. The remaining 6% of the clones showed no identifiable match to AAUAAA. There are a variety of possible explanations for the existence of these clones, including 3' end formation by an alternative mechanism, such as trans-splicing of a downstream gene in a polycistronic transcription unit, for instance.
It is clear from the tables shown below that certain mismatches are tolerated, namely any change in position 1 and a G in position 4. It is noteworthy that the 2-base mismatches are all combinations of the most frequent mismatches at these two positions. We feel confident in concluding that most of the sequences that appear in the correct locale more than about 1% of the time are actually serving as cleavage and poly(A) addition signals in C. elegans, since no other options appear to be available in these cases. However, it should be noted that those that appeared very rarely could have done so by a chance match to the functional sequence. Hence, sequences with mismatches in positions 5 or 6, for example, may not actually represent functional AAUAAA's.
The bar graph shows the locations of the sequences identified as poly(A) signals in this survey. In most cases there are between 11 and 15 nucleotides between the 3' end of the signal and the beginning of the A stretch. Note that because we don't have the genomic sequence for most of the cDNAs used here, the actual number of bases between the signal and the site of addition of unencoded A's could be somewhat higher, because the site of addition could be an A or even a series of A's in a significant number of cases.
To summarize our results: C. elegans uses the same signal to specify the site of cleavage and poly(A) addition as do vertebrates: AAUAAA. However, in contrast to vertebrates, in C. elegans only half the genes use a perfect match to this consensus. Tolerated mismatches were found in another 44% of the genes, while in 6% of the sequences poly(A) signals were not identified. In general AAUAAA and its variants occurs about 13 nucleotides upstream of the poly(A). It has been observed previously, that AAUAAA can be present in the 3' untranslated region of a C. elegans mRNA and not result in cleavage just downstream. Our analysis confirms that perfect AAUAAA's (as well as acceptable mismatches) are often ignored. This suggests that additional sequences are involved. In vertebrates these additional sequences have been shown to be U-rich or G+U-rich stretches with no obvious consensus. We are now looking to see whether we can identify an analogous sequence in C. elegans. However, since this sequence is expected to be downstream of the cleavage site, the next search must be done in a genomic library.
