Worm Breeder's Gazette 12(2): 49 (January 1, 1992)
These abstracts should not be cited in bibliographies. Material contained herein should be treated as personal communication and should be cited as such only with the consent of the author.
The tra-1 gene is both necessary and sufficient for female somatic development in C. elegans. We recently described (WBG vol. 11, no. 5) the characterization, cDNA cloning, and sequencing of transcripts from the tra-1 region. The tra-1 region encodes two mRNAs, one of 1.5 kb found primarily in L2 ,and one of 5 kb, found at all stages. Both mRNAs can encode proteins containing zinc finger motifs, two fingers in the case of the shorter RNA and five in the case of the longer. The fingers are very similar to those of the human putative oncogene GLI and the Drosophila segment polarity gene ciD.
In order to prove that at least one of the two mRNAs we detected in the tra-1 region really does encode tra-1 ,we have sequenced cDNA from tra-1 amber mutants. Eight amber mutants have been isolated (Hodgkin, 1987) and we sequenced cDNA from seven of these. The cDNA was generated by PCR amplification of reverse transcribed RNA from 20-100 homozygous mutant animals carrying an amber suppressor mutation. Sequencing was by PCR linear amplification (Craxton, 1991). Using these methods it is possible to go from worm to cDNA sequence in a single (long) day.
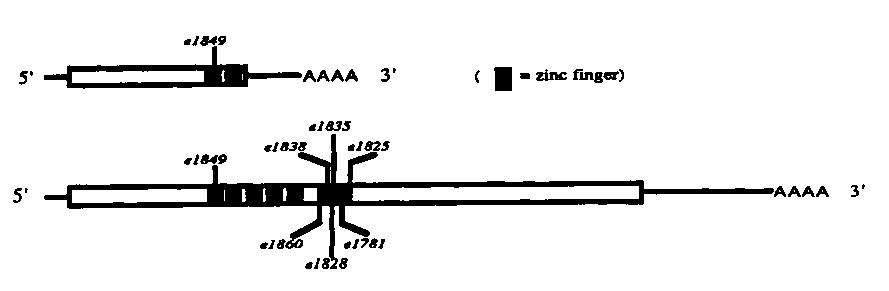
The seven tra-1 amber mutations are indeed in the two putative tra-1 RNAs, as diagrammed below:
[See Figure 1]
In general, the stronger mutations are 5' to the weaker ones, and with only two exceptions ( e1838 and e1860 ),the order is the same as that derived from crude intragenic mapping experiments (Hodgkin, 1987). Interestingly, six of the amber mutations lie in a region of only 180 bp (indicated by the black box). Since 30% of the potential amber sites (mainly CAG glutamine codons) of the protein are in this region, this is perhaps not surprising. What is surprising is the ratio of genetic to physical map distances in the region. For example, the genetic map distance between e1825 and e1828 is very approximately 0.1 mu (Hodgkin, 1987), while the physical distance is only 93 bp. Intriguingly, the region contains nine copies of a heptamer that resembles repeats found in mammalian recombinational hotspots and which does not occur elsewhere in the transcript.
Since mutants that remove most or all of the boxed region are more severe than those that leave most or all of it, the region may be important either to stabilize what is left of the protein or for interactions with other proteins. The region is predicted to form an alpha helix, and, in a helical wheel projection, forms a curious-looking structure with alternating glutamine and alanine residues. Whether this is at all significant is not clear.
It might be expected that the amber mutants that truncate the protein after the zinc fingers would not prevent DNA binding and might therefore cause a dominant negative phenotype under the right conditions. This appears not to be so: animals that are heterozygous for the amber mutations and homozygous for the smg-2 mutation e2008 (to increase the amount of amber product) do not show any dominant negative phenotype (JH, unpublished).
Most of the amber mutants were isolated by reversion of a dominant gain-of function mutant e1575 .We found that both the amber mutants and the parent e1575 allele have an asparagine to aspartate mutation near the amino terminus. There are no other mutations in the transcribed region 5' to the amber mutations ( e1575 clearly maps to the 5' side of all the ambers) so this mutation is likely to be responsible for the e1575 phenotype. It may therefore define a site of negative regulation, possibly by one of the fem genes (though it could of course be an extraneous second mutation). We are currently sequencing other gf alleles to see if they also have alterations in this region.
Hodgkin, J. (1987) Genes Dev. 1:731-745.
Craxton, M (1991). in Methods: a Companion to Methods in Enzymology 3, pp. 20-26, Academic Press.