Worm Breeder's Gazette 10(2): 57
These abstracts should not be cited in bibliographies. Material contained herein should be treated as personal communication and should be cited as such only with the consent of the author.
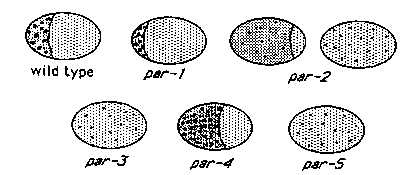
Cortical actin distribution in one-cell embryos from hermaphrodites homozygous for mutations in any of the 5 par genes is different from that found in N2 wild type embryos. par mutants are maternal effect lethal mutations characterized by defects in early cleavage patterns, P-granule localization and cleavage timing (Kemphues, et al. 1988. Cell 52: 311-320). . Since actin microfilaments appear to play a significant role in cytoplasmic localizations (Hill and Strome, 1988. Dev. Biol. 125: 75-84), we decided to examine the distribution of microfilaments in early embryos of par mutants . During the time from fertilization to first cleavage, the arrangement of cortical actin fibers undergoes a dramatic, but transient, change in wild type embryos (Strome, S., 1986. J. Cell Biol. 103: 2241-2252). This change can be seen by fluorescence microscopy when embryos are lightly fixed with formaldehyde and stained with rhodamine-labeled phalloidin. Just after fertilization, filaments and small actin foci appear evenly distributed on the embryo's cortex. During pronuclear migration, actin distribution becomes asymmetrical: large, brightly staining foci gradually appear in the anterior cortex of the embryo, forming a brightly staining 'cap' which covers a third to a half of the embryo. Concurrently the posterior portion of the embryo loses most of its actin foci leaving fine, lightly staining filaments. At about the time of pronuclear meeting, anterior actin accumulation is maximal. During metaphase, the actin foci seem to disperse and the asymmetry disappears leaving actin filaments and small foci evenly distributed on the embryo's surface by the end of anaphase. Cortical actin staining patterns found in embryos from hermaphrodites homozygous for par mutations differ from this wild type pattern in a gene specific manner. The differences are most evident around the time of pronuclear meeting and metaphase when the N2 'cap' is most pronounced and are summarized in Fig. 1 and as follows: 1) par-1 (b274 and it32). Anterior actin caps are present, and brightly staining; however, they are generally smaller and more compact than those of N2. 2) par-2 (it53 and it49). Actin caps either are not seen at all, or are weakly staining and large, covering 2/3 or more of the embryo. 3) par-3 (it62 and it54). Cortical actin is symmetrically distributed throughout the one-cell stage and caps are not found. 4) par-4 (it33, it47 and it57). Actin caps are brightly staining but cover half to two-thirds of the embryos. In addition, preliminary data suggest that the cap at metaphase is somewhat smaller than that at pronuclear meeting; whereas in N2 the metaphase cap is generally somewhat larger than that found at pronuclear meeting. 5) par-5(it55). Actin caps cannot be detected in any stage of these embryos. These results are consistent with the notion that the actin microfilaments are integral components of the machinery involved in early cytoplasmic localizations or that their distribution pattern results from the same system responsible for cytoplasmic localizations in one-cell embryos. On the other hand, abnormal cortical actin patterns may be common among mutants with early developmental defects. We are currently investigating this possibility by looking at cortical actin patterns in embryos from mutants with defects in genes that appear not to be directly involved in cytoplasmic localization. Staining Procedure: One and two cell embryos from N2 animals or from animals with mutations in one of the 5 par genes were cut out of hermaphrodites into water and mounted onto polylysine coated slides. ( Very sticky slides can be prepared by placing a drop of a 1 mg/ml polylysine solution on an ethanol washed slide and rubbing the polylysine onto the slide with a clean latex rubber Pasteur pipet bulb. ) Embryos were covered with a solution of formaldehyde (2.4% formaldehyde in 0.108M potassium phosphate, pH 7.4, 0.2% Triton X-100); slides were mounted on an inverted microscope equipped with Nomarski optics and a long distance working lens; and vitelline membranes were immediately cracked by gentle pressure from a glass needle attached to a microinjection apparatus. Embryos were fixed for 20 minutes and washed with 2 changes of 0.125M potassium phosphate buffer, pH 7.4, for at least 20 minutes. Embryos were incubated with rhodamine- labelled phalloidin (6 units/ml) in 0.12M potassium phosphate, pH 7.4, 0.2% Triton X-100, for 20 minutes and rinsed in phosphate buffered saline (PBS). Chromosomes were stained with DAPI (1x10+E-4 mg/ml for 1 min.) and rinsed in PBS for 1 min to 2 hr. Fixed embryos were mounted for fluorescence microscopy in 1 mg/ml phenylenediamine in 80% glycerol, 20% PBS, pH 8.5. [See Figure 1]